使用AIQM1表征环对苯撑纳米套索大型分子
Published Time: 2023-05-29 09:56:24
通过点击化学反应将供电子、电中性、吸电子芳基取代基引入炔基嵌入的[11]环对苯撑([11]CPPs)中对其进行功能化得到一系列纳米套索分子。我们使用最先进的人工智能增强量子力学方法1(AIQM1)对这些化合物及其与富勒烯的复合物的电子和光物理性质进行了全面分析。开箱即用的AIQM1能够快速、准确地对这些数百个原子组成的大型分子体系进行计算,进而更好地理解和解释实验中观察到的现象。
Alkyne-embedding [11]cycloparaphenylene ([11]CPPs) was functionalized with electron-donating, -neutral, and -withdrawing aryl substituents to yield a series of nanolassos via click chemistry. We used our state-of-the-art, artificial intelligence-enhanced quantum mechanical method 1 (AIQM1) to thoroughly analyze the electronic and photophysical properties of these compounds and their complexes with fullerenes. Out-of-the-box AIQM1 allowed performing fast and accurate calculations to better understand and explain the phenomena observed experimentally for these big systems with hundreds of atoms.
在最近的工作中,我们的合作者通过无铜[3+2]叠氮-炔点击化学,用一系列供电子、电中性和吸电子的芳基取代基对具有应变的嵌入炔烃[11]CPP进行功能化。这种功能化导致了如下图所示的套索状结构。
In our recent work, our collaborators functionalized strained alkyne-embedding [11]CPP with a series of electron-donating, -neutral, and -withdrawing aryl substituents through copper-free [3+2]azide-alkyne click chemistry. This functionalization resulted in lasso-shaped structures shown below.
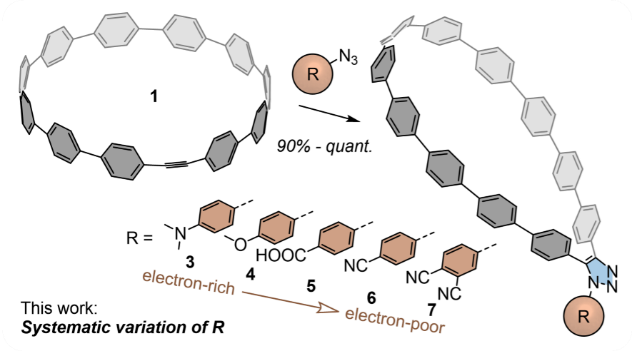
实验合作者们还通过X射线晶体学分析了它们的结构,并用核磁共振光谱、稳态和瞬态吸收和发射光谱以及电化学等技术表征和分析了它们的电子和光物理性质。由于上述化合物的形状可以轻松地捕获富勒烯分子,因此,我们还研究了纳米套索-富勒烯复合物。
The collaborators also analyzed these molecular nanolassos by X-ray crystallography and their electronic and photophysical properties were probed with NMR spectroscopy, steady-state and transient absorption and emission spectroscopies as well as electrochemistry. The shape of the above compounds allows us to easily capture fullerenes and our study investigated nanolosso-fullerene complexes too.
我们的团队通过理论计算来解释所观察到实验现象的本质。纳米套索及其与富勒烯的复合物都非常大,由数百个原子组成,对它们进行高水平计算的成本非常高昂。因此,为了确保计算的高质量,我们主要使用了AIQM1方法,该方法比常用的DFT方法更快且更准确。
Our group performed theoretical calculations to explain the nature of observed experimental phenomena. The nanolassos and their complexes with fullerenes are rather large with up to several hundreds of atoms, thus, it is computationally expensive to perform high-level calculations on them. Thus, to ensure the high quality of the calculations, we mostly used our AIQM1 approach which is both faster and more accurate than popular DFT approaches.
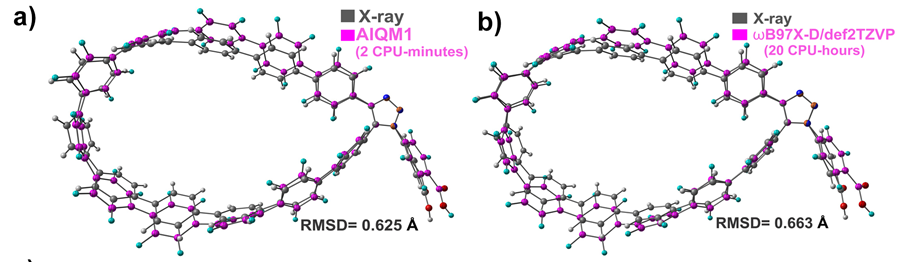
为了验证AIQM1方法在这种规模的化合物中的可用性,我们对其中一种化合物比较了AIQM1优化构型与X射线晶体结构。如下图所示,两种几何结构之间的均方根位移(RMSD)非常小(约为0.63 Å),其中的差异也可能是AIQM1气相优化中未考虑堆叠效应造成的。相比之下,DFT优化耗时更长(20 CPU小时;AIQM1为2 CPU分钟),而且精度也不高(RMSD为0.66 Å)。
To verify the use of AIQM1 for compounds of this size, we compared AIQM1-optimized geometry with X-ray crystallographic structure of one of the compounds. As can be seen from the figure below, the root mean square displacement (RMSD) between two geometries is quite small (ca. 0.63 Å) with differences that may be also caused by the packing effects not considered in our gas-phase optimizations. For comparison, DFT optimization is both much more time-consuming (20 CPU-hours compared to 2 CPU-minutes at AIQM1) and not more accurate (RMSD of 0.66 Å).
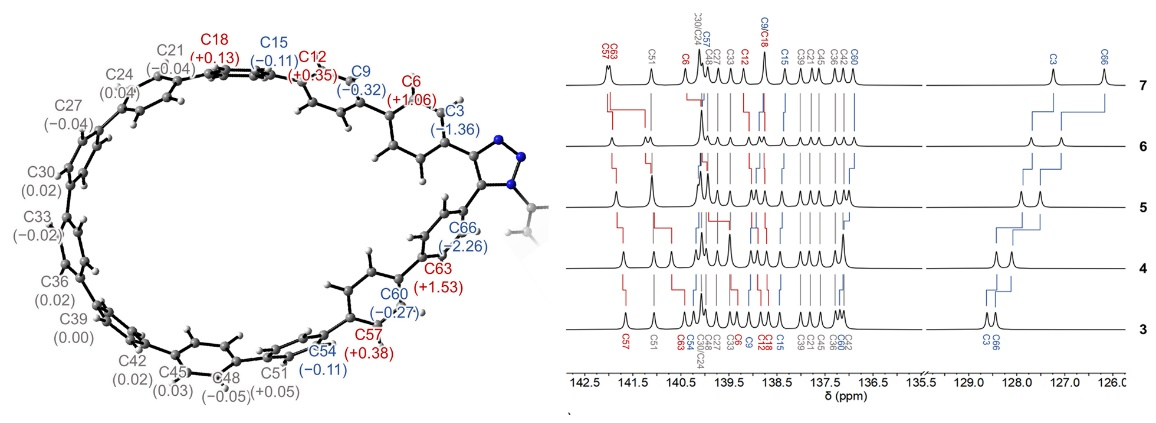
通过在AIQM1优化的几何结构上进行的DFT 13C NMR位移计算,我们可以将实验观察到的峰值明确地指定给特定的原子,并显示随着取代基的吸电性增加,Cipso原子的位移朝低场偏移。AIQM1计算还可以用于研究纳米套索化合物的氧化和还原性质,计算结果不仅遵循了取代基的预期趋势,也揭示了氧化和还原时的电荷分布情况,也即从完全到部分电子/空穴定域在碳环/取代基上。
Theoretical 13C NMR shifts calculated with DFT on AIQM1-optimized geometries allow to unambiguously assign experimentally observed peaks to specific atoms and show downfield shifts for the Cipso atoms with increasing electron-withdrawing properties of the substituents. AIQM1 calculations of oxidation and reduction properties of nanolassos not just followed the expected trend for substituents but also revealed the charge distribution upon oxidation and reduction, which ranges from full to partial electron/hole localization on the carbon loop/substituent.
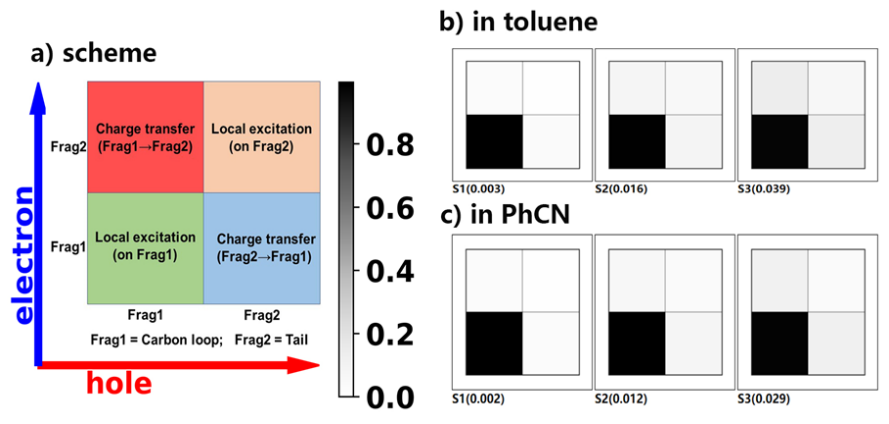
其中一个具有挑战性的重要观察结果是溶剂对纳米套索化合物吸收和荧光光谱的影响:光谱在极性溶剂中呈现轻微的红移。一种猜测是由于可能存在的分子内电荷转移引起的,但我们在AIQM1优化的几何结构上进行的TDDFT计算以及随后的单电子密度矩阵(1TDM)分析并未显示出碳环和取代基之间的电荷转移,而是表明激发主要局域在碳环上(弗伦克尔激子)。这一令人困惑的现象被解释为溶剂对基态几何结构微妙但重要的影响。
One of the key observations which was challenging to explain is the solvent effect on the absorption and fluorescence of nanolassos: The spectra are slightly red-shifted in more polar solvent. One could guess that it may be due to the plausible intramolecular charge transfer, but our TDDFT calculations on the AIQM1-optimized geometries and subsequent one-electron density matrix (1TDM) analysis did not reveal any charge transfer between carbon loop and substitute and shows that excitations are localized on the carbon loop (Frenkel excitons). This puzzling phenomenon was explained by subtle but important effect of solvent on geometry in the ground state.
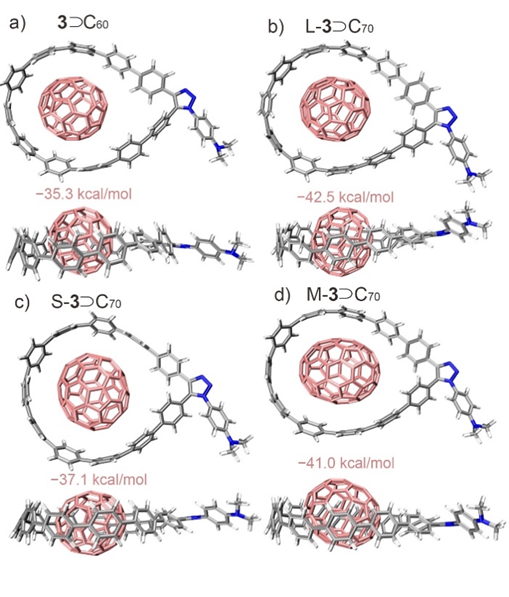
对于纳米套索与富勒烯的络合,我们发现与C60相比,套索分子与C70的结合能力更强,因为C70的椭圆形状与CPP衍生物的套索形状更匹配。这一点通过我们的AIQM1计算和我们合作者的实验质谱测量得到了证实。然而,从实验中,我们无法确定C70相对于碳环的准确排列方式:它可能是平躺(L-)的、竖立(S-)的或半躺半立(M-)的。通过AIQM1,我们可以轻松地分析并发现最稳定的构型是平躺的,见下图所示的几何形状和结合能。
Turning to the complexation of nanolassos with fullerenes, we found that complexation with C70 is stronger than to C60as the oval shape of C70 is a better match to the lasso-shape of CPP derivatives. This is confirmed by our AIQM1 calculations and our collaborators’ experimental mass-spectrometry measurements. From the experiment, however, we could not judge how exactly is C70 aligned relative to the carbon loop: it could be in lying (L-) or standing (S-) or in half-lying, half-standing (M-). With AIQM1, we could easily analyze and find that the most stable configuration is lying, see the Figure below for geometries and binding energies.
此外,我们利用AIQM1进行了组态相互作用单重态(CIS)计算,以优化自由纳米套索和其与富勒烯的复合物在第一激发态中的几何结构,并计算其荧光。我们的AIQM1/CIS计算表明,自由纳米套索的荧光应该在复合物形成后被猝灭,这确实是实验所观察到的。
Importantly, configuration interaction singles (CIS) calculations with AIQM1 allowed use to optimize geometries of free nanolassos and their complexes with the fullerenes in the first excited state and calculate fluorescence. Our AIQM1/CIS calculations showed that fluorescence of free nanolassos should be quenched upon complexation and this is indeed what was observed experimentally.
我们的工作清楚地表明,人工智能量子增强的化学方法已经可以准确快速地进行有机化合物复杂现象的常规研究。值得注意的是,以上所有计算都是使用开箱即用的AIQM1进行的,没有进行任何重新训练等操作,也即像使用任何其他典型的量子化学方法一样。
Our work clearly demonstrates that AI-enhanced quantum mechanical methods can be already routinely used to investigate complex phenomena in organic compounds with high accuracy and speed. Noteworthy, all above calculations were done with the out-of-the-box AIQM1 without any retraining, etc., i.e., as one would use any other typical quantum chemical methods.
AIQM1是开源免费的,详细的教程可以在 MLatom.com/AIQM1上找到,并且可以通过我们的MLatom@XACS云计算服务在Web浏览器上进行基本的AIQM1计算。
AIQM1 is open-source and free to use with detailed tutorials on its use available at MLatom.com/AIQM1 and basic calculations with AIQM1 can be performed free of charge via a web browser using our MLatom@XACS cloud computing service.
原文链接 / Link:
Tobias A. Schaub*, Anna Zieleniewska, Ramandeep Kaur, Martin Minameyer, Wudi Yang, Christoph M. Schüßlbauer, Lina Zhang, Markus Freiberger, Lev N. Zakharov, Thomas Drewello, Pavlo O. Dral, Dirk Guldi, Ramesh Jasti*. Tunable Macrocyclic Polyparaphenylene Nanolassos via Copper‐Free Click Chemistry. Chem. Eur. J. 2023, e202300668, Early View. DOI: 10.1002/chem.202300668. (blog post)