使用(p)KREG模型构建精准分子势能面
Published Time: 2023-07-27 19:20:08
为了更精确地描述分子势能面,我们在(p)KREG模型的公式中考虑了梯度信息。测试结果显示,(p)KREG模型的精度优于或等同于其它最新机器学习模型。此外,我们发现,同时学习能量和梯度对于构建势能面至关重要(仅学习能量或梯度无法满足需求)。
We improved (p)KREG models for an accurate representation of molecular potential energy surfaces (PESs) by including gradient information explicitly in their formalism. Our models are better or on par with other state-of-the-art machine learning models as we show on extensive benchmarks. We also found that learning both energy and energy gradients are important to properly model potential energy surfaces and learning only one of them is not enough. The methods and benchmarks are reported in the J. Chem. Theory Comput.. Code in MLatom and tutorials are available online and calculations can be performed on the XACS cloud.
机器学习拟合多维函数的能力非常强大,可用于拟合诸如势能面、激发能、偶极矩和电子密度等性质。拟合的关键在于选择分子描述符编码分子的几何和/或其它信息。核方法(例如核岭回归),是该领域中最广泛使用的机器学习算法之一(其它的方法还有神经网络和线性模型)。
Machine learning is very powerful in fitting multi-dimensional functions. In quantum chemistry, this capability of ML is exploited to fit such properties as potential energy surfaces, excitation energies, dipole moments, and electron densities. The key is also the choice of descriptors that encode geometrical and/or other information about molecules. The kernel methods (KM) such as kernel ridge regression (KRR) are among the most widely used machine learning algorithms in this field, together with neural networks and linear models.
我们之前开发了一些学习量子化学性质的机器学习方法,包括基于KRR的KREG模型,其使用了全局描述符RE(relative-to-equilibrium,由归一化的原子间距离获得)和高斯核函数。pKREG模型是KREG模型的衍生模型,使用了交换不变核函数以保证原子交换不变性。
One of our previously developed ML approaches to learning quantum chemical properties is the KRR-based KREG model which uses a global relative-to-equilibrium (RE) descriptor based on scaled internuclear distances and the Gaussian kernel function; its pKREG variant is based on a permutationally invariant kernel applied to enforce invariance with atom permutations.

我们之前的工作发现,(p)KREG模型能够高效且准确地学习标量性质(势能、激发能、振子强度(http://mlatom.com/mlatom-1-2-ml-absorption-spectra/))。如果在核方法的公式中加入导数信息可以极大地提高模型的准确性,因此在本工作中,我们在(p)KREG模型中加入了显式学习导数的功能。
Our previous works showed that (p)KREG models can be used to efficiently and accurately learn scalar properties (potential energies, excitation energies, oscillator strengths). However, it is known that the accuracy can be greatly improved by including derivative information explicitly in the KM formalism. Thus, in this work, we implement the explicit learning of derivatives in (p)KREG models.
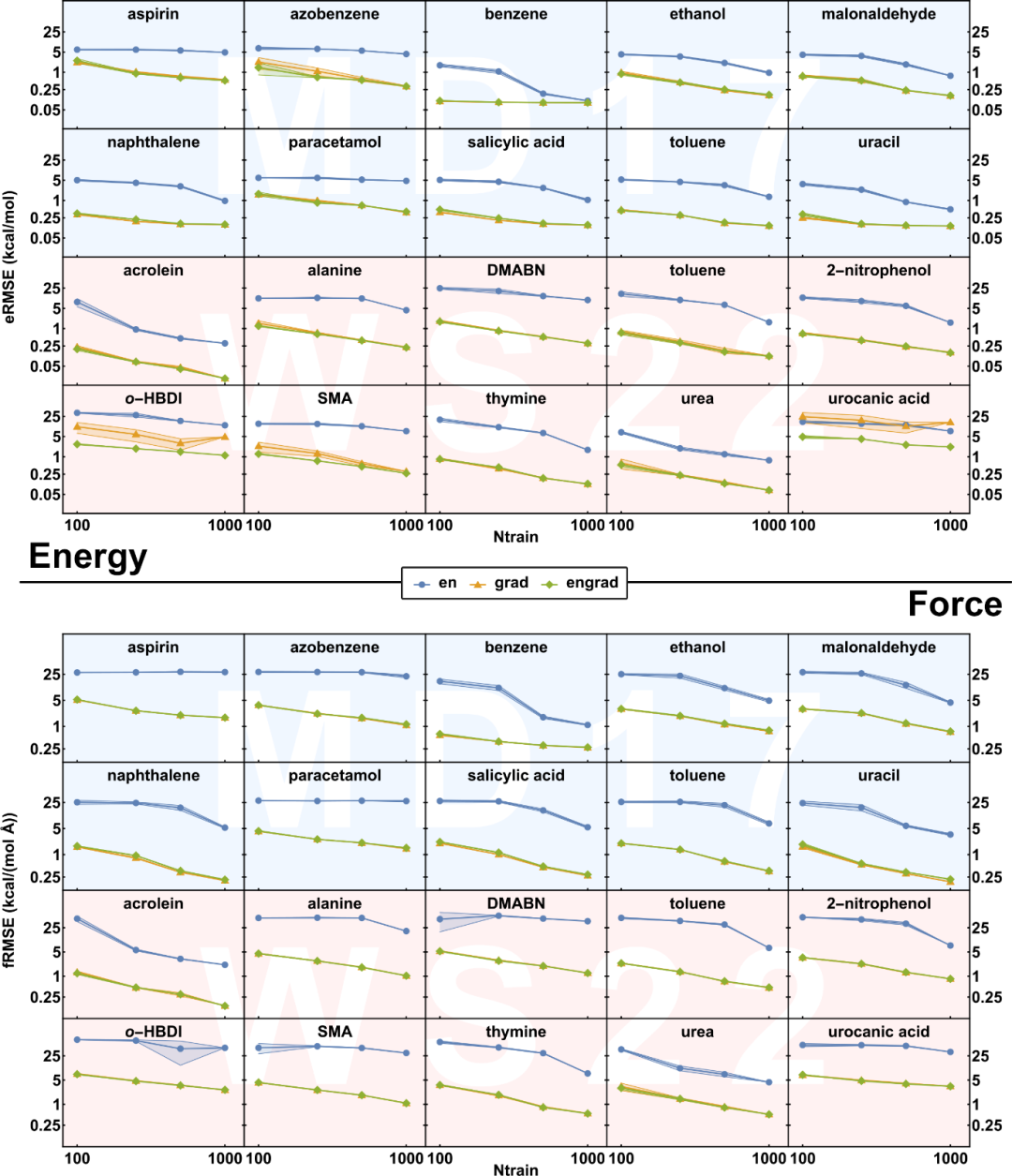
我们在MD17和最新构建的WS22数据集上测试了(p)KREG模型学习单分子势能面的表现。从下面KREG模型的学习曲线中可以看到,学习能量梯度信息能极大地提高模型的性能(与预期一致)。此外,同时学习能量和梯度让模型更稳定地学习更具挑战性的分子势能面(如WS22中的尿刊酸(urocanic acid,其单独用能量梯度模型无法准确模拟势能面))。因此,若能量和能量梯度都可用,我们建议对两者进行训练,因为加入能量不会显著增加计算时间,却可以极大提高模型在小数据集上的稳定性。
We evaluated the performance of our models on single molecule potential energy surfaces of the popular MD17 dataset and our recently developed, more challenging WS22 dataset. From the learning curves of KREG models below, we can see that including energy gradient information is necessary to significantly improve the performance of models, as expected. Besides, including both energies and energy gradients makes the model more robust for some challenging cases such as urocanic acid in the WS22 dataset, where models trained only on energy gradients fail completely. As a result, we recommend training on both energies and energy gradients if both are available as including energies does not bring much additional computational cost but greatly improves the model robustness in small data settings.
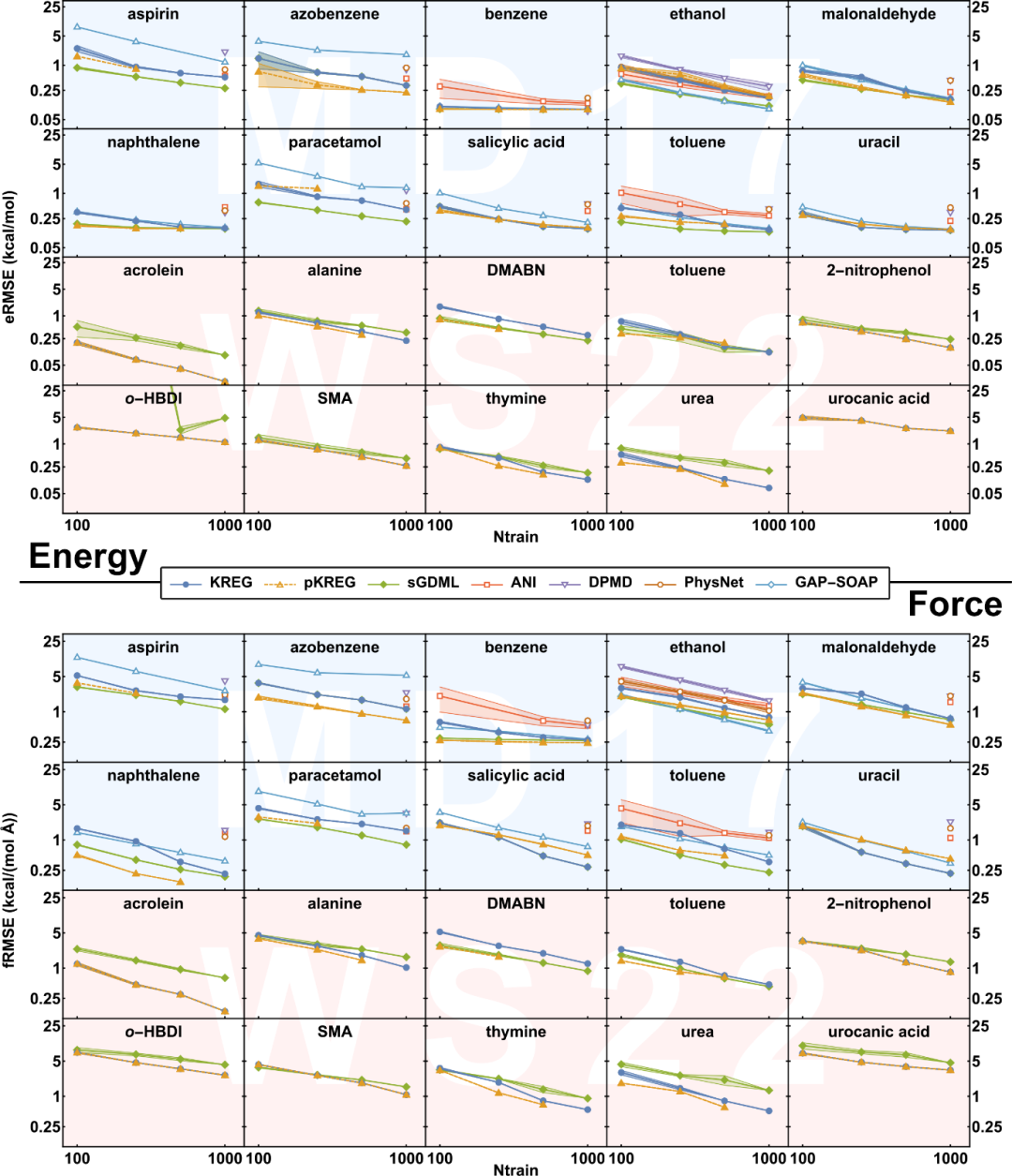
我们还将(p)KREG模型与其它最新的机器学习模型进行比较,如sGDML、ANI、DPMD、PhysNet和GAP-SOAP。从学习曲线中可以看到,对于所有势能面,pKREG模型的准确性和其它模型相差无几。对于小训练集和一些简单且能量分布较窄的分子(比如MD17中的分子),KREG模型的表现较差,pKREG模型的表现更好(与sGDML相当)。当训练集变大时,KREG与pKREG之间的差距会变小,这可能是由于大训练集中潜在包含分子的对称信息。对于WS22数据集中能量分布较宽且结构更扭曲的分子,以及在两个数据集中使用较大训练集时,(p)KREG模型的表现会逐渐超过sGDML。最后,对于更具挑战性的分子如urocanic acid和o-HBDI,sGDML没有显式学习能量,所以预测能量时的表现很差。
We also compare our (p)KREG models with other state-of-the-art machine learning models such as sGDML, ANI, DPMD, PhysNet and GAP-SOAP. The learning curves show that pKREG model has competitive accuracy across all potential energy surfaces. For small training sets, and some simple molecules and narrower distribution of energies (such as in MD17), KREG underperforms, while pKREG has higher accuracy, which is comparable to sGDML. The difference between KREG and pKREG becomes smaller as the size of the training set becomes larger, which is caused by the underlying symmetry information in larger training sets. For broader energy distributin and more distorted structures as in the WS22 dataset and for larger training sets in both MD17 and WS22 datasets, (p)KREG models start to have better performance than sGDML. Finally, for some challenging cases like urocanic acid and o-HBDI, sGDML, which does not learn energies explicitly, fails to learn energies.
KREG和pKREG模型代码和相应教程均可在开源软件包MLatom中获取,也可以在XACS云计算平台上免费使用。
Both KREG and pKREG models can be accessed in our open-source package MLatom, which can also be used on the XACS cloud computing platform free of charge at MLatom@XACS. Tutorials can be found at http://mlatom.com/kreg/.
论文信息:
Yi-Fan Hou, Fuchun Ge, Pavlo O. Dral. Explicit Learning of Derivatives with the KREG and pKREG Models on the Example of Accurate Representation of Molecular Potential Energy Surfaces. J. Chem. Theory Comput. 2023, 19 (8), 2369–2379. DOI: 10.1021/acs.jctc.2c01038