超越3D机器学习原子间势:迎接4D时空原子级人工智能模型
Published Time: 2023-09-01 10:32:57
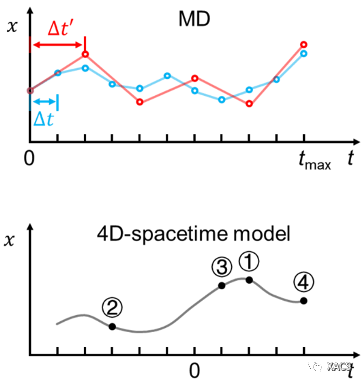
我们引入了一个概念,即:使用四维(4D)时空原子人工智能(AI)模型学习分子随时间的变化。我们通过开发4D时空GICnet模型证明这个概念是可行的。与传统动力学不同的是,该模型可以直接预测分子的原子坐标以作为时间的连续函数,而传统分子动力学是在离散的时间步长上,通过缓慢连续、一次一步的算法来预测核坐标。
We have introduced a concept of 4D-spacetime atomistic AI models (https://pubs.acs.org/doi/10.1021/acs.jpclett.3c01592) that learn how the molecule changes in time. We demonstrate that this concept is feasible by developing the 4D-spacetime GICnet models that directly predict the atomic coordinates of a molecule as a continuous function of time. It is a radically different alternative to traditional molecular dynamics that predicts nuclear coordinates with slow consecutive, one-step-at-a-time, algorithms at discrete time steps.
分子始终在运动中,模拟其时间相关的动态行为是很重要的。但是,精确的动力学是一项非常耗费资源的工作,通常需要进行许多近似处理。其中最常用的近似之一是分子动力学(MD),即:计算在一个非常小的时间步长内原子的移动量,并多次重复此过程直至达到给定时间。上述MD每步计算都需要评估作用在原子上的力,但计算力并不是一个简单的任务,目前通常使用所谓的机器学习原子间势来加速。然而,这种机器学习方法仍然无法避免要在离散时间步长上运行缓慢连续算法的需要。
The molecules are always in motion and it is highly desirable to simulate their time-dependent, dynamic, behavior. Unfortunately, accurate dynamics is very resource-consuming endeavor and it is often done with many approximations. One of the most widely used approximations is molecular dynamics which calculates how much atoms are moved in a very small time step and then repeats this procedure many times until the required time is reached. Each of these steps requires the evaluation of forces acting on atoms. The problem is that calculating forces is not a trivial task but it is now often accelerated with so-called machine learning interatomic potentials. Nevertheless, such ML approaches still do not obliterate the need of running slow sequential algorithms with discrete time step.
人工智能在科学中的进步使我们产生了一个新的想法:是否可以直接用人工智能预测给定时间内的原子位置?当时我们正在撰写一篇关于AI量子动力学方法的论文(至今已过去很长一段时间),该方法将量子动力学轨迹作为时间的连续函数进行学习。最终,我们于2021年10月29日的会议上决定开展研究原子位置随时间变化的工作。
Advances in AI for science allow us to look at the problem from a radically new angle: why not predict the atomic positions for the required time directly with AI? This was an idea floating around in our group for quite some time and, back then, we had been working on a paper on the related AI-quantum dynamics (http://dr-dral.com/predicting-the-future-of-excitation-energy-transfer-in-light-harvesting-complex-with-artificial-intelligence-based-quantum-dynamics/) approach that also learns quantum dynamics trajectories as a continuous function of time. We finally set to work on learning atomic positions as a function of time in our meeting on October 29, 2021.

2021年10月29日,我们开始建立4D时空AI模型。从右到左:Pavlo、Fuchun(4D-GICnet模型的主要开发者)、Yi-Fan(传统MD代码、红外和功率谱模拟的开发者)、Arif(相关的AI量子动力学方法的开发者)、Peikun(AI量子力学方法1的开发者)、Lina(基态动力学方法的主要用户和测试者,专长为非绝热动力学)。Xinxin后来也加入该小组,并从一个反应的4D-GICnet模型中提出了科学见解(其研究兴趣是将机器学习应用于化学反应)。
29 October 2021, when we set to work on the 4D-spacetime AI models. From right to left: Pavlo, Fuchun (the main developer of 4D-GICnet models), Yi-Fan (developer of traditional MD code, IR and power spectra simulations), Arif (developer of the related AI-quantum dynamics approach:http://dr-dral.com/predicting-the-future-of-excitation-energy-transfer-in-light-harvesting-complex-with-artificial-intelligence-based-quantum-dynamics/), Peikun (developer of the AI-quantum mechanical method 1:http://dr-dral.com/artificial-intelligence-makes-accurate-quantum-chemical-simulations-more-affordable/), Lina (main user and tester of ground-state dynamics approaches and a specialist in nonadiabatic dynamics:https://www.sciencedirect.com/science/article/abs/pii/B9780323900492000081?via%3Dihub). Xinxin joined the group later and contributed with extracting insight from the 4D-GICnet models for a reaction (her research interest is application of ML to reactions:http://dr-dral.com/evaluating-aiqm1-on-reaction-barrier-heights/).
我们可以证明,对于最简单的双原子系统,机器学习基本上可以预测任何时间点的分子构型。然而,对于乙醇或偶氮苯等多原子分子,维度的限制会产生影响。为解决该问题,我们专门开发了更通用的GICnet模型,该模型能够预测较长时间(10-20飞秒,远超过MD中使用的典型时间步长(如1飞秒))内的核坐标。
We could show that for simplest systems like diatomics, the realization of the concept (https://pubs.acs.org/doi/10.1021/acs.jpclett.3c01592) is rather straightforward and ML basically can predict the configuration at any time. However, for polyatomic molecules such as ethanol or azobenzene, the dimensionality curse kicks. For such molecular systems, we developed more general GICnet model that is able to predict the nuclear coordinates up to rather distant time: 10-20 fs. This time is much longer than typical time steps used in molecular dynamics (e.g., preferably below 1 fs).
通过使用GICnet模型,我们可以比传统的MD算法(即使已使用机器学习原子间势加速过)更快地获得分子轨迹,可并行预测不同时间步长的轨迹(因为它们不需要按顺序计算),所预测轨迹可以实现任意高的分辨率,也可以快速计算能从MD轨迹中推导出的性质(我们通过生成振动光谱来说明)。
One can use the GICnet models to obtain molecular trajectories much faster than with traditional MD algorithms (even accelerated with machine learning interatomic potentials) and we can parallelize predictions at different time steps as they do not need to be calculated sequentially. The trajectories also can be obtained with arbitrarily high resolution. This allows to also calculate fast the properties which can be derived from MD trajectories and we demonstrate this by generating vibrational spectra.
除更快的MD算法外,GICnet模型中的神经网络凝结了分子在4D时空中的相关信息,可用于深入了解耦合核运动(其相互作用的分析难度较大)。我们通过分析不同坐标对偶氮苯的顺反异构化的影响对此加以说明。
The GICnet models are of course much more than simply faster MD algorithm. The neural networks condense the information about a molecule in 4D spacetime. This can be used, e.g., to get insights into the coupled nuclear motions whose interplay are otherwise difficult to interpret. For the demonstration, we analyzed how different coordinates affect the cis-trans isomerization of azobenzene.
动力学模拟从传统MD转向4D时空AI模型将是大势所趋。为了降低准入门槛,我们在开源MLatom中提供了上述模型,相关模拟工作也可以在MLatom@XACS云计算平台上进行。
The future will surely see the shift of dynamics simulations from traditional MD to 4D-spacetime AI models. To reduce the entry barrier, we provide our models in open-sourceMLatom (https://github.com/dralgroup/mlatom/tree/gicnet) and the simulations can be also run on MLatom@XACS cloud (https://xacs.xmu.edu.cn/).
Paper:
Fuchun Ge, Lina Zhang, Yi-Fan Hou, Yuxinxin Chen, Arif Ullah, Pavlo O. Dral. Four-dimensional-spacetime atomistic artificial intelligence models. J. Phys. Chem. Lett. 2023, 14, 7732–7743. DOI: 10.1021/acs.jpclett.3c01592.
Preprint on ChemRxiv: https://doi.org/10.26434/chemrxiv-2022-qf75v(AncientOriginalVersion)| arXiv: https://doi.org/10.48550/arXiv.2308.11311 (more recent version).