Inorg. Chem. : Application of chemical bond analysis in lithium-sulfur batteries design
Published Time: 2024-04-07 15:38:03
Editor's note: New example on the application of XACS showcases the study of Wang Changwei from Shaanxi Normal University and Mo Yirong from the University of North Carolina at Greensboro. The physical factors influencing the conformational preferences of lithium polysulfide clusters in Lithium-sulfur batteries were elucidated utilizing energy decomposition analysis method based on the BLW approach, combined with GKS-EDA calculations in the XEDA@XACS software package. This study tends to offer a new theoretical perspective for the rational and efficient design of lithium-sulfur batteries.

Structures are of fundamental importance for diverse studies of lithium polysulfide clusters, which govern the performance of lithium-sulfur batteries. The ring-like geometries (Figure 1) were regarded as the most stable structures, but their physical origin remains elusive. Notably, the lithium polysulfide clusters have intricate conformational space and diverse intermolecular interactions, making systematical analysis at theoretical level essential in this study.
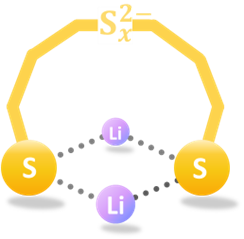
Figure 1. Ring-like Structure of Li2Sx (x = 4−8).
An insightful understanding of the
conformational preferences can be obtained by incorporating bonding analysis
methods into a comprehensive study of all low-lying isomers. In this regard,
the ab initio valence bond (VB) theory stands out as an ideal option. As the
simplest ab initio VB method, the block-localized wave function (BLW) retains
the localized orbitals but simplifies the multireference wave function to a single
determinant, making it computationally efficient. Its associated energy
decomposition (BLW-ED in short) approach can decompose the binding energy into
physically meaningful energy components and has provided a multitude of new
insights into the conventional and unconventional interactions.
In this work, we combined ABCluster program with BLW-ED and GKS-ED method to explore the conformational preferences of Li2Sx (x = 4−8) clusters. In details, the lowest-lying structure and its isomers, which lie within 50.00 kcal/mol higher than the most stable one were retained. Key factors for the conformational preferences were explored by examining the contributions of individual energy components to the relative stabilities with references to the most stable isomers. The energy decomposition analysis results of Li2S8 clusters in Figure 2 indicate that polarization stabilization governs the relative stabilities of most low-lying isomers.
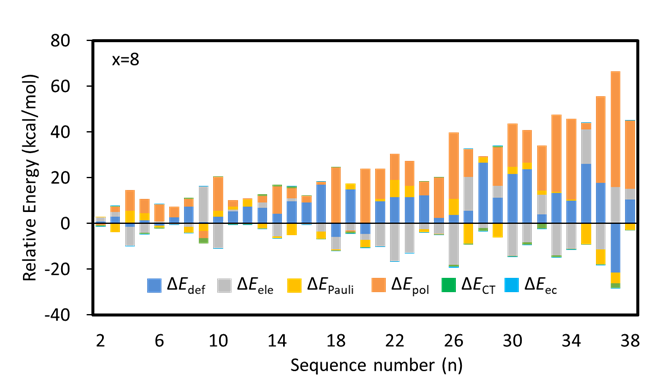
Figure 2. Relative values of all energy components in the energy decomposition analysis for Li2S8.
To understand the geometric correlations with the polarization energy term, we explored various structural parameters and eventually identified the Li+···Li+ distance as the principal index for polarization. Specifically, a close arrangement of the two cations leads to an enhanced resultant electric field, which ultimately exerts an enhanced force for the polarization within the Sx2− dianion (Figure 3a). Figure 3b shows that the polarization energy beautifully correlates with its cooperative component linearly. This suggests that the relative strength of polarization is primarily governed by the cooperative effect. Furthermore, the cooperative components of both total interaction and polarization energies also exhibit a linear correlation (Figure 3c), indicating that the overall cooperative effect is predominantly dominated by polarization interactions. In summary, the conformational preference of Li2Sx (x = 4−8) clusters mainly originates from the cooperative effect of the polarization interaction, which is strengthened by the closely located Li+ cations within the quadrilateral binding region of the ring-like geometries.
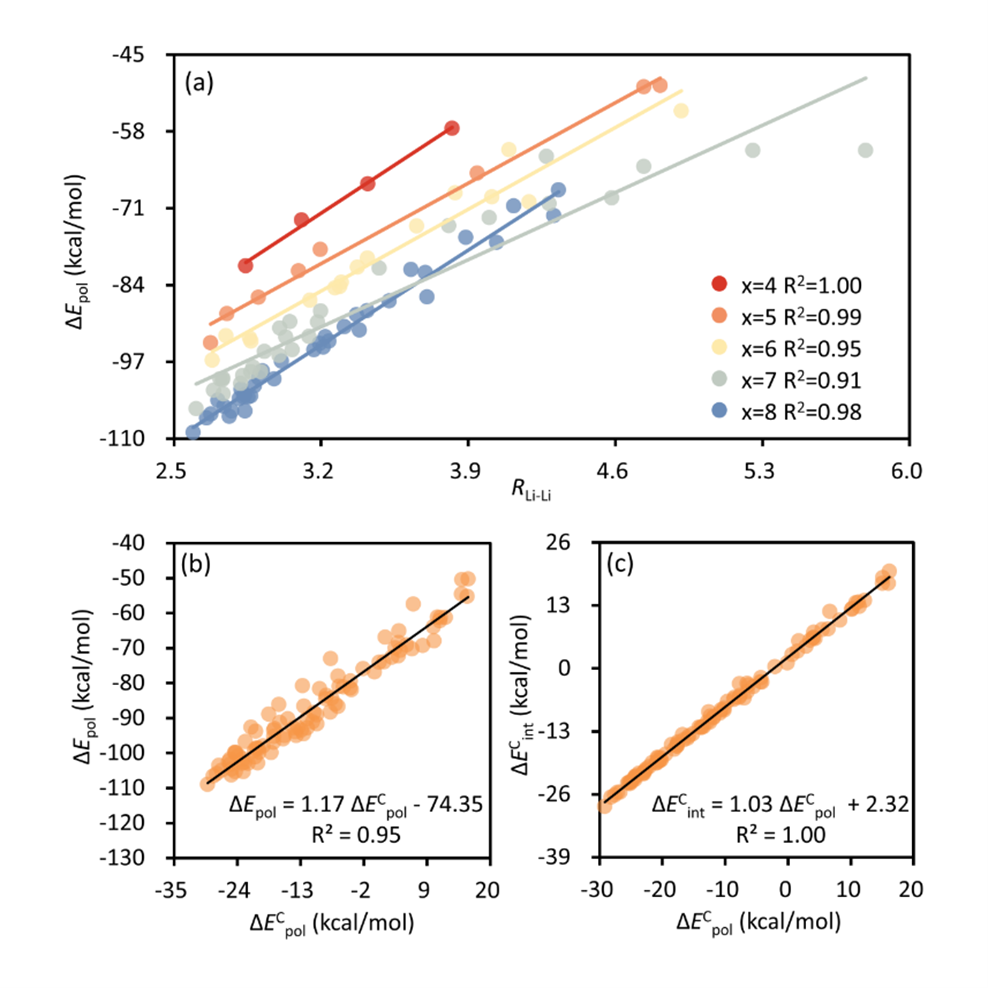
Figure 3. (a) Correlation between the polarization energy and the Li···Li distance for all low-lying isomers of each Li2Sx (x = 4−8) cluster.
(b) Correlation between the polarization energy and its cooperative component and (c) correlation between cooperative components of total interaction and polarization energies of all isomers.
This work utilized the XEDA program in the XACS platform to calculate the electrostatic, Pauli exchange repulsion, and electron correlation components in energy decomposition analysis, which is crucial for analyzing the interactions and conformation preference in lithium polysulfide clusters.
Paper:
Conformational Preference of Lithium Polysulfide Clusters Li2Sx(x=4–8) in Lithium–Sulfur Batteries
Xinru Peng, Jiayao Li, Jingshuang Dang, Shiwei Yin, Hengyan Zheng, Changwei Wang*, and Yirong Mo*
XACS Research Highlights
XACS Research Highlights are posts about examples of research carried out using the XACS platform. We welcome the users to submit their blog posts to us at xacs@xmu.edu.cn or directly to one of the PIs and we will highlight your research on the official XACS channels and social accounts. We reserve the right to small editorial changes for clarity and corrections.