Nat. Commun.: Accelerating reliable multiscale quantum refinement of protein-drug systems enabled by AIQM1
Published Time: 2024-08-01 14:50:13
Recently, the Chung group at Southern University of Science and Technology (SUSTech) has combined efficient machine learning potentials (MLPs) with multi-scale quantum refinement methods to enhance computational efficiency and reliability. Several general-purpose MLPs (e.g. AIQM1, ANI-1ccx, ANI-2x) were used to replace quantum mechanics (QM) methods and to refine 50 different protein-drug/inhibitor structures. MLPs give reliable structures compared to those using ωB97X-D methods as the QM method. Additionally, two levels of MLPs (CC and DFT-quality) were combined for the first time to overcome MLP limitations. Furthermore, computational benchmark datasets (PB20-QM, PB20-QM-8 k and PB20-QM-3 k) were established, which hopefully help future development of better DFT, MLPs and/or SE methods for drug/inhibitor molecules. These research results were recently published in Nature Communications (DOI: 10.1038/s41467-024-48453-4). The CC-quality AIQM1 method in MLatom@XACS software was used and yielded the best results for the protein-drug/inhibitor structures (particularly those containing charged groups).
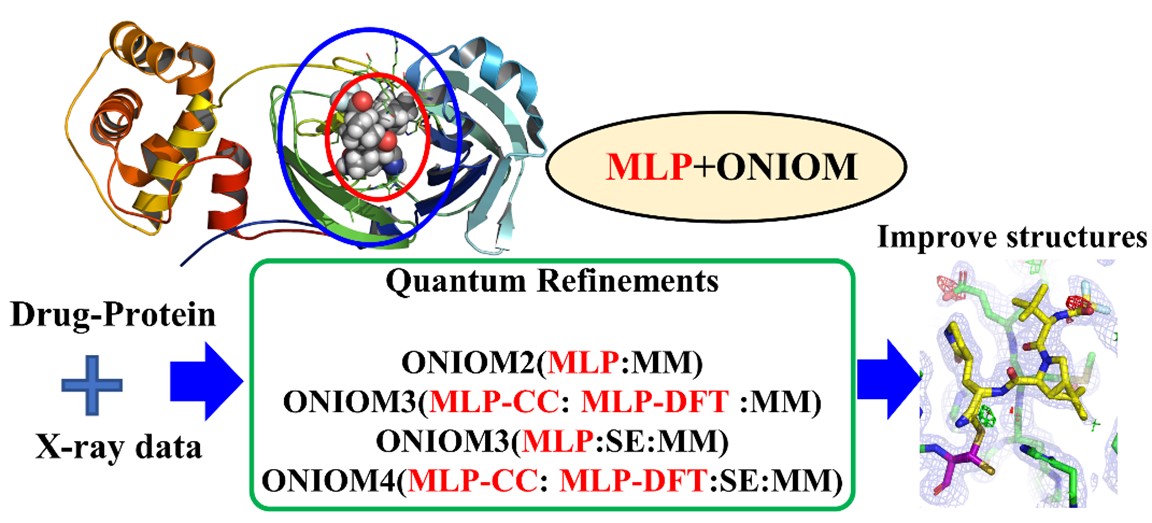
Fig. 1. Computational approaches
Accurate atomic structures of biomacromolecules are vital for drug development and biocatalysis. X-ray diffraction (XRD) has long been one of the most common methods to determine the atomic structures of numerous biomacromolecules. Structural determination often relies on standard X-ray crystallographic refinement methods, in which the molecular mechanics (MM) force field is combined with experimental (XRD) data to derive reasonable chemical structures. On the other hand, Ryde and coworkers pioneered development of quantum refinement, which replaces the quick MM method with more accurate quantum mechanics (QM) methods to describe the key site in proteins and improve the local structures (J. Comput. Chem. 2002, 23, 1058). In addition, other groups have developed QR method by combining multiscale, linear-scale QM, fragmentation and quantum-embedding methods to further boost or improve the refinement process. The Chung group at SUSTech also proposed to combine multi-scale ONIOM method with QR method (ONIOM_QR), which successfully improved the structural accuracy for the key local metal binding site in metalloproteins (J. Chem. Theory Comput. 2021, 17, 3783). However, the extensive computational costs for multi-scale QM/MM, QM/SE or QM/SE/MM methods (especially high-level quantum-embedding CCSD-in-DFT as the QM method) hinder the broad applications of QR to many biological systems.
In this study, the Chung group further incorporates robust machine learning potentials (MLPs) in multiscale QR method (ONIOM_QR) to improve refinement efficiency and reliability (Figs. 1 & 2a). First, computational benchmark datasets (PB20-QM, PB20-QM-8 k (mainly containing C, H, O, N, F, S, Cl elements) and PB20-QM-3 k (mainly containing C, H, O, N elements)) were established to evaluate the structural reliability of 3–19 k drug/inhibitor molecules computed by the DFT, MLPs and SE (GFN2-xTB) methods (Fig. 2c). To apply refinement of drug/inhibitor molecules containing more elements and to overcome MLP limitation, two different levels (CC- and DFT-quality) of MLPs or MLP+xTB were further combined for the first time through an extrapolative ONIOM approach, such as ONIOM2(ANI-1ccx:ANI-2x), ONIOM2(AIQM1:ANI-2x), ONIOM2(MLP:xTB). In such approach, MLP becomes the high level in the ONIOM method to treat the core drug/inhibitor structures (Fig. 2b). Then, this unique MLPs+ONIOM-based QR method was applied to successfully refine 50 different protein–drug/inhibitor systems. Based on RSZD scores, strain energy, bond distance, angle and rotatable dihedral results, MLPs can give reliable results (Fig. 2d). Compared to ANI series, the CC-quality AIQM1 method gives higher accuracy, particularly challenging systems containing charged groups.
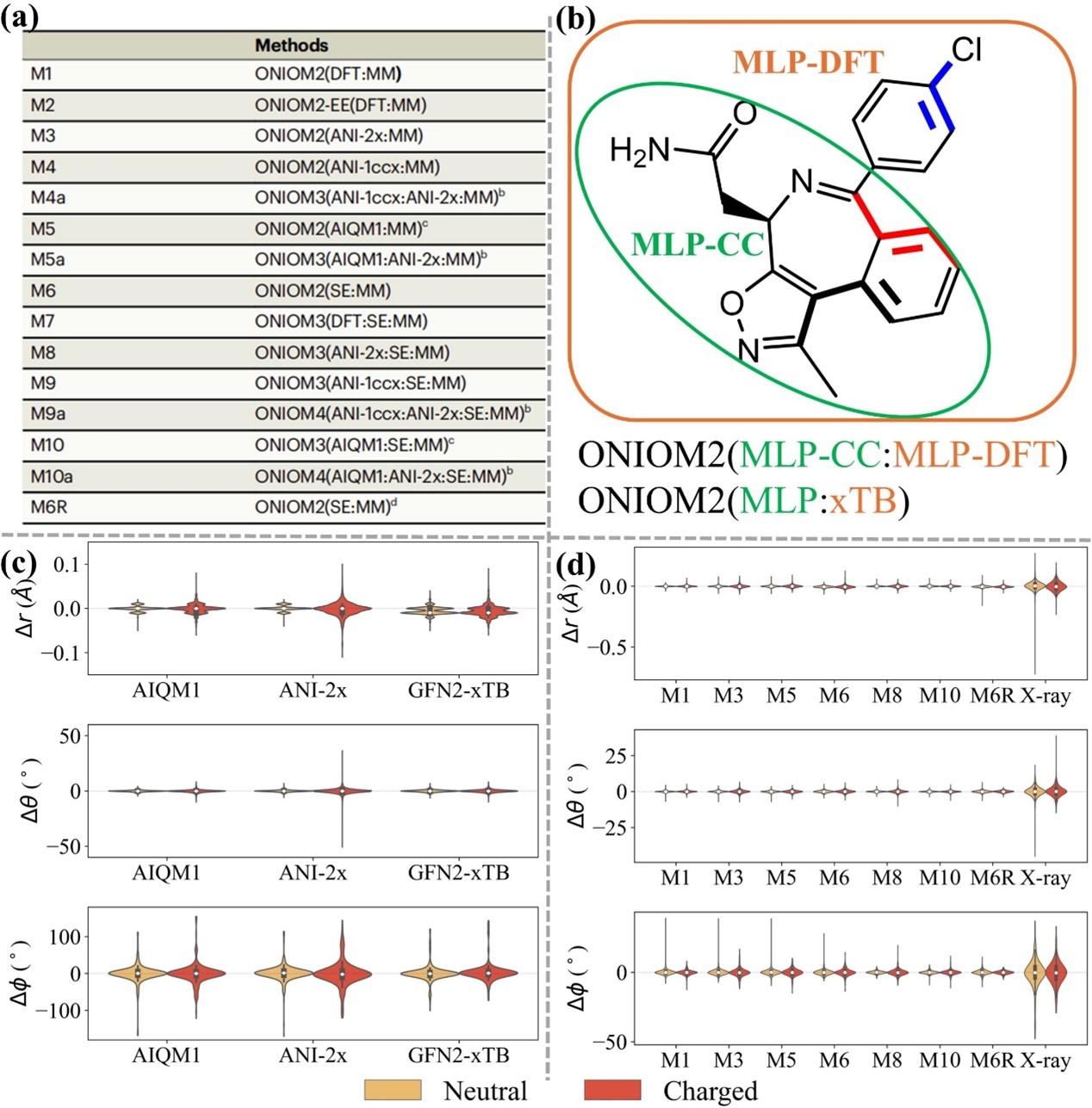
Fig. 2 (a) Different computational chemistry methods in the quantum refinements; (b) Combination of MLPs and xTB by ONIOM approach; The refined bond distance, angle and rotatable dihedral results for 50 protein–drug/inhibitor systems by different computational schemes (c) in gas phase or (d) the protein.
Moreover, quantum refinement (QR) on one crystal structure of wild-type SARS-CoV-2 main protease with nirmatrelvir (PDB ID: 7RFW) shows the possible existence of both bonded and nonbonded conformers between nirmatrelvir and Cys145 with a possible ratio of these conformers (~7:3, Fig. 3). The refined results suggest that such occupation ratio gives the greatest structural improvement, which should provide some structural insights for the design of more effective SARS-CoV-2 drugs.
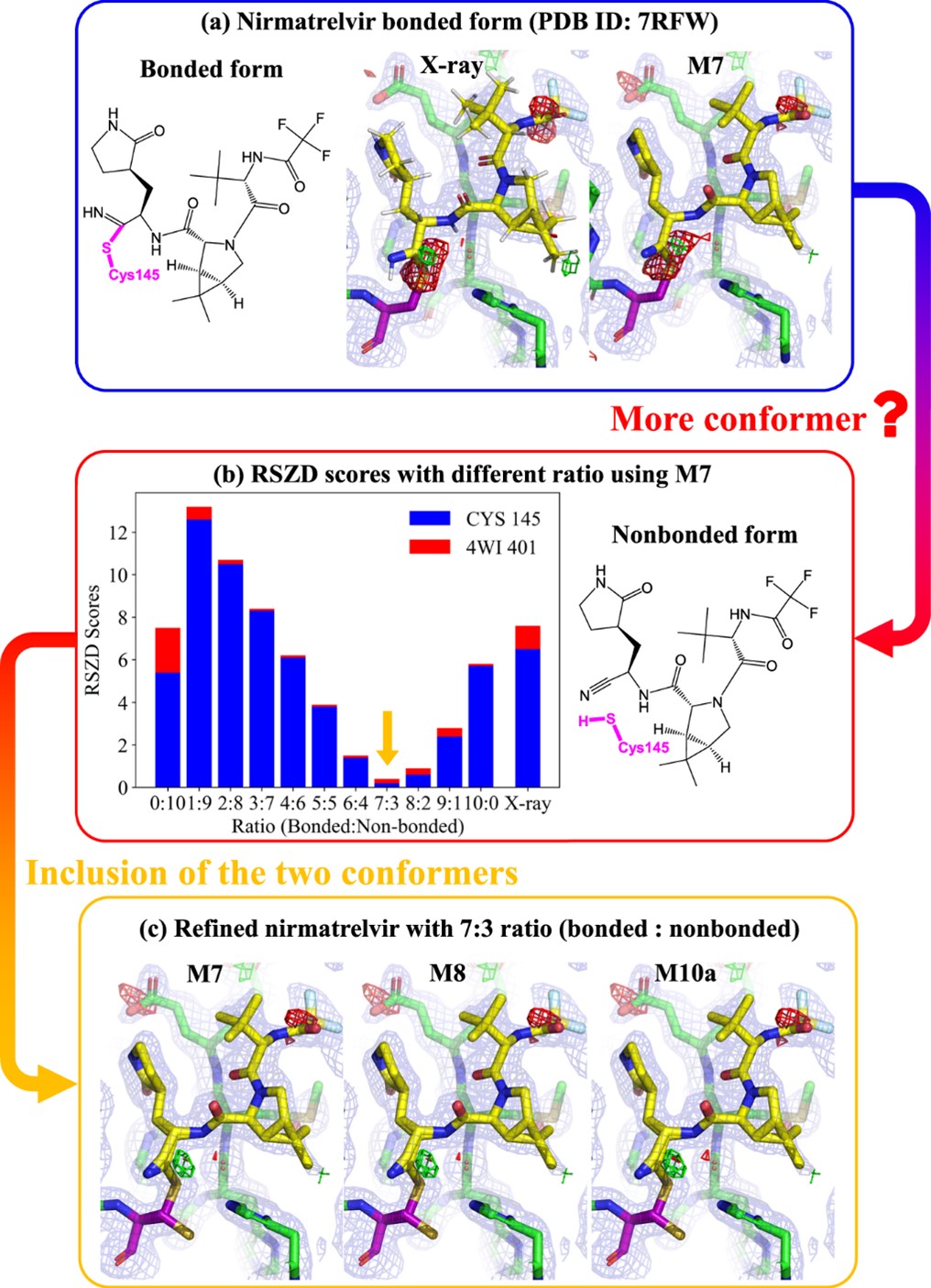
Fig. 3. One wild-type SARS-CoV-2 main protease crystal structure with nirmatrelvir contains two possible conformers: (a) bonded and (b) nonbond forms. (c) The electron density maps after quantum refinement on the nirmatrelvir binding site in the SARS-CoV-2 main protease refined by various quantum refinement schemes and including the two conformers.
Apart from X-ray crystallography, MLPs is potentially helpful for modern structural determination methods of biomacromolecules (e.g. Cryo-EM, MicroED). Furthermore, computational benchmark datasets (PB20-QM, PB20-QM-8 k and PB20-QM-3 k) were established to evaluate the structural reliability of 3–19 k drug/inhibitor molecules computed by the DFT, MLPs and/or SE methods, which hopefully help future development of better DFT, MLPs and/or SE methods for drug/inhibitor molecules.
Paper:
Zeyin Yan, Dacong Wei, Xin Li & Lung
Wa Chung*
Nat.
Commun. 2024,
15, 4181. DOI:
10.1038/s41467-024-48453-4