J. Phys. Chem. A丨从头算价键分子动力学:SN2反应机理研究
Published Time: 2025-06-23 15:36:23
在化学反应动力学领域,从头算分子动力学(AIMD)模拟一直是揭示反应机理的重要工具。价键理论作为一种重要的电子结构方法,能够为化学键和反应机理提供清晰直观的化学图像。然而,由于价键理论中非正交轨道的使用,能量梯度的计算一直是一个难题。本文基于约化密度矩阵(RDM)算法,开发了VBSCF方法的高效能量梯度算法,成功克服了这一难题,并首次将从头算经典价键理论与分子动力学模拟相结合,提出了从头算价键分子动力学(AIVBMD)方法,为化学反应动力学的研究提供了新的工具。 为了验证AIVBMD方法的有效性,本文以气相中的经典SN2反应(F⁻ + CH₃Cl → CH₃F + Cl⁻)为例进行了模拟。研究结果表明,在相同的活性空间下,VBSCF方法仅需27个价键结构即可在定性上准确描述SN2反应的势能面,而基于分子轨道的CASSCF方法则需要5292个组态函数(CSFs)。此外,AIVBMD方法通过分析价键结构权重的变化,清晰地揭示了反应过程中C-Cl键的断裂、C-F键的形成以及短暂出现的氢键,为反应机理提供了直观的化学图像。
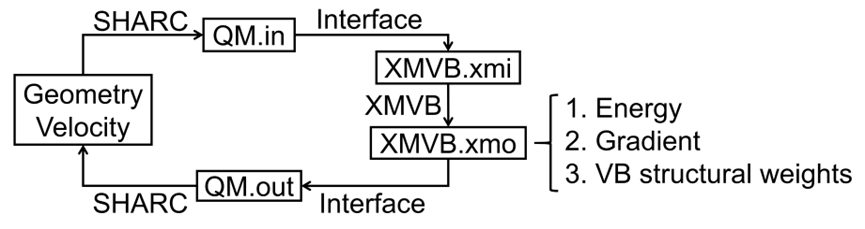
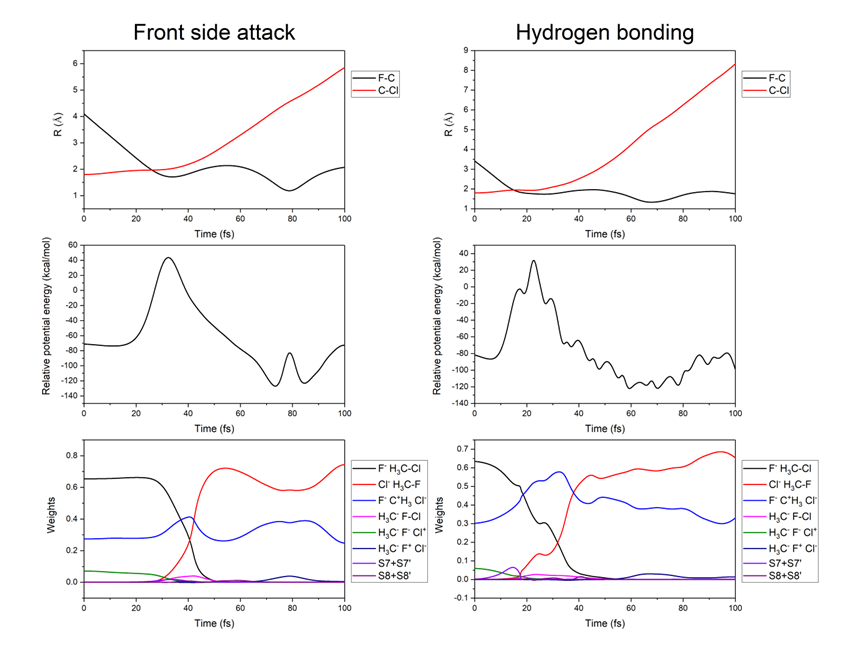
本文通过将从头算经典价键理论与分子动力学模拟相结合,成功发展了AIVBMD方法。在相同活性空间下,VBSCF方法相比CASSCF方法仅需使用少量的价键结构即可在定性上准确描述势能面,为动力学研究提供了一种基于紧凑多组态波函数的计算方案。同时,通过研究价键结构权重在反应过程中的演化,AIVBMD方法能够揭示化学键断裂与形成的详细信息,为反应机理提供了直观的理解。该工作展示了价键理论在化学反应动力学研究中的巨大潜力,为后续的动力学研究奠定了坚实基础。
论文信息:Ab Initio Valence Bond Molecular Dynamics: A Study of SN2 Reaction Mechanisms
Miao Guo, Xun Wu, Wei Wu,* and Chen Zhou*
DOI: 10.1021/acs.jpca.4c08431