AIQM2
AIQM2 is the second generation of our AI-enhanced quantum mechanics methods (AIQM) series. Its high speed, competitive accuracy, and robustness enable organic reaction simulations beyond what is possible with the popular DFT methods. It can be used for transition state (TS) structure search and reactive dynamics, often with chemical accuracy. Except for energies, AIQM2 can also provide other molecular properties, e.g., dipole moment for generating infrared (IR) spectra.
More details can be found in our recent publication on Chemical Science:
Yuxinxin Chen, Pavlo O. Dral*. AIQM2: Organic Reaction Simulations Beyond DFT. Chem. Sci. 2025, accepted manuscript. DOI: 10.1039/D5SC02802G. Preprint on ChemRxiv: https://doi.org/10.26434/chemrxiv-2024-j8pxp (2024-10-08).
The blog post for an overview of AIQM2 is also online.
AIQM2 is already available in our open-source MLatom software. It is also one of the most frequently used methods on the Aitomistic Hub for online simulations via a web browser, including the AI assistant Aitomia.
Although AIQM2 is limited to the CHNO elements, AIQM2@UAIQM is available for all but the 7th row elements. AIQM2 is available via PyPI and on GitHub, AIQM2@UAIQM as described in the UAIQM tutorial.
Prerequisites
If you want to try AIQM2 locally, the following packages are required:
MLatom (https://github.com/dralgroup/mlatom, it’s recommmended to use the latest version of MLatom where we improved many functionalities of using AIQM2. The MLatom version should be greater than 3.12.0)
DFT-D4 (https://github.com/dftd4/dftd4, please use the pypi version and find the executable
dftd4under/binfolder of the environmnet)
After the installation of DFT-D4, you need to set the environment variable dftd4bin=[your DFT-D4 binary]
As an alternative, you can directly try AIQM2 on our online platform Aitomistic Hub without the need for environment installation. We will show how to run AIQM2 on Aitomistic Hub in the following tutorial. The required scripts are the same if you want to run locally.
Your first try on Aitomistic Hub
On Aitomistic Hub, you can check the molecule you want to simulate within the visualization window.
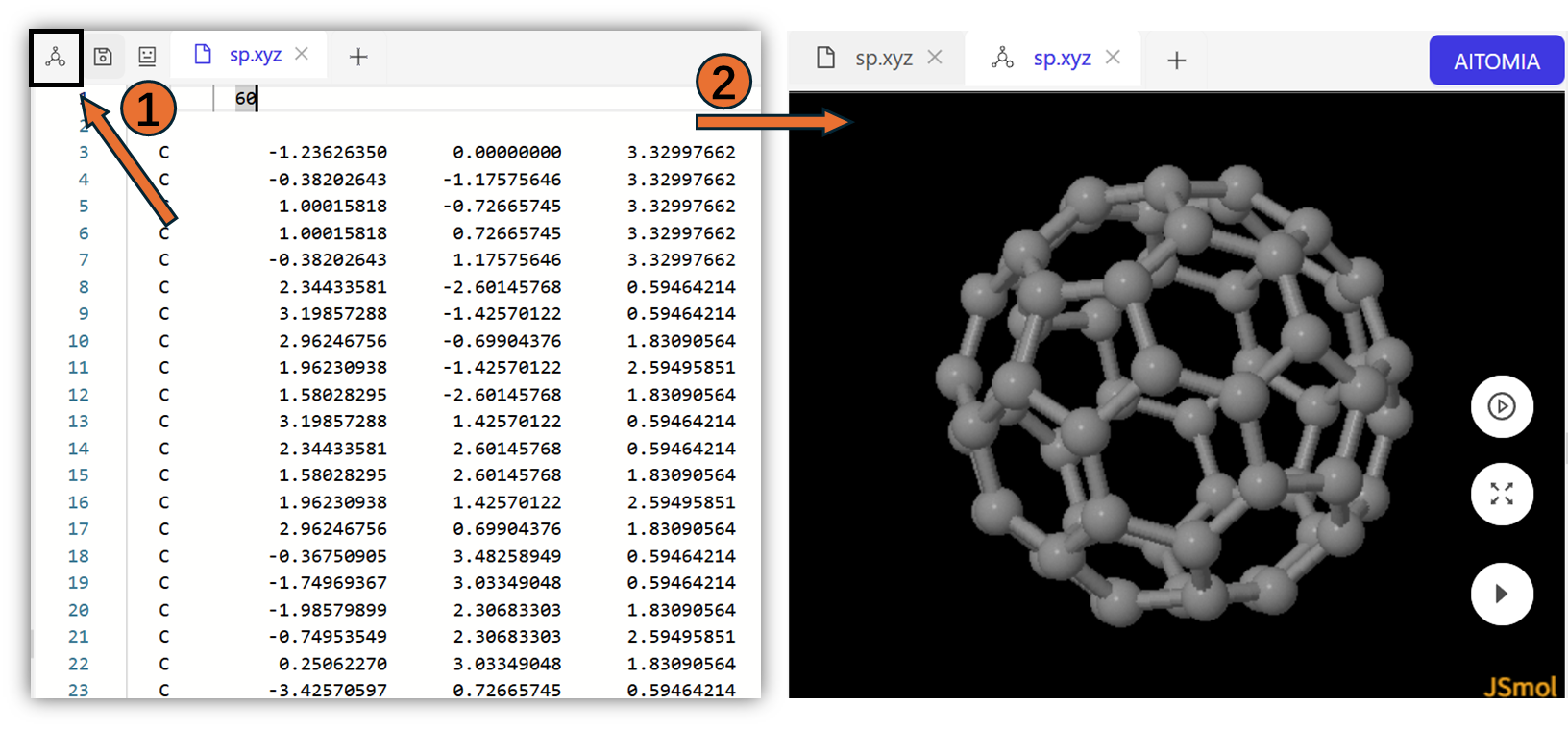
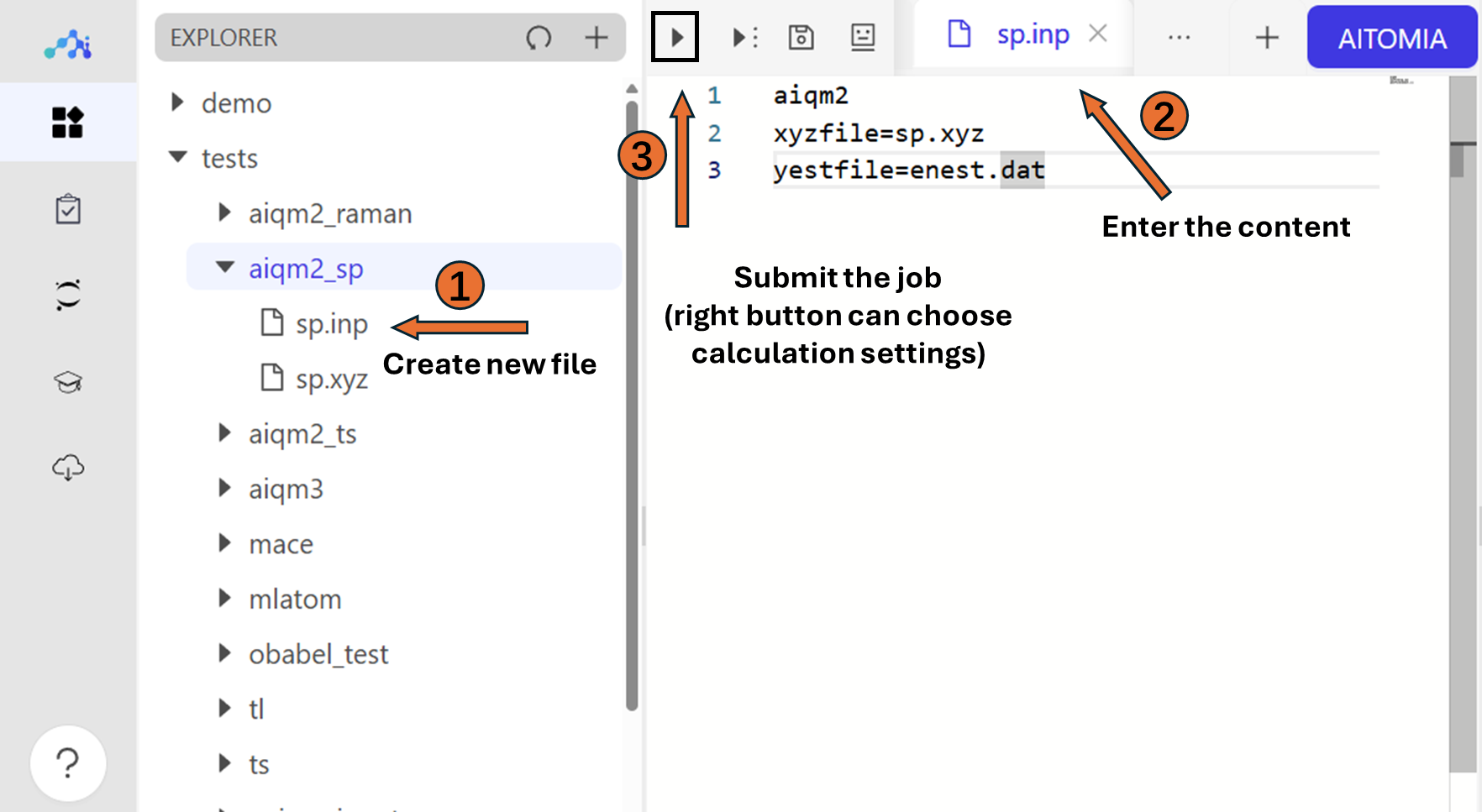
To perform single point calculation with AIQM2, the required input file follows:
AIQM2
xyzfile=sp.xyz
yestfile=enest.dat
You can click the submit button on the left top corner to submit the job. After several seconds, there will be .out file in the current directory.
Another way to use AIQM2 is via python script. You can launch the jupyter notebook on the left panel and start coding:
import mlatom as ml
mol = ml.data.molecule.from_xyz_file('sp.xyz')
aiqm2 = ml.models.methods(method='AIQM2')
aiqm2.predict(molecule=mol, calculate_energy=True, calculate_energy_gradients=True, calculate_hessian=True)
print(mol.energy)
print(mol.get_energy_gradients())
print(mol.hessian)
Detailed tutorials for single point calculations can be found here.
Transition state optimization and validation
For input file, you can try the following scripts:
aiqm2
ts
freq
xyzfile=init.xyz
This input file will perfrom transition state geometry optimization first ,and then perform frequency calculation to get the normal modes and the corresponding frequencies. The optimized geometry file can be found in optgeoms.xyz. You can use Chemcraft to visualize each normal mode with the generated .log file.
The detailed tutorial about using MLatom for geometry optimization can be found here and frequency calculations here. We also have dedicated section for transtion state.
Reactive molecular dynamics with AIQM2
This part resembles that in UAIQM tutorial where AIQM2 is selected from the library based on the requested time budget.
IR spectrum with AIQM2
AIQM2 uses its baseline GFN2-xTB to get dipole moment and its derivatives for generating IR spectrum. You can find the detailed tutorial here. The basic usage via input file follows:
ir
AIQM2
xyzfile=opt.xyz
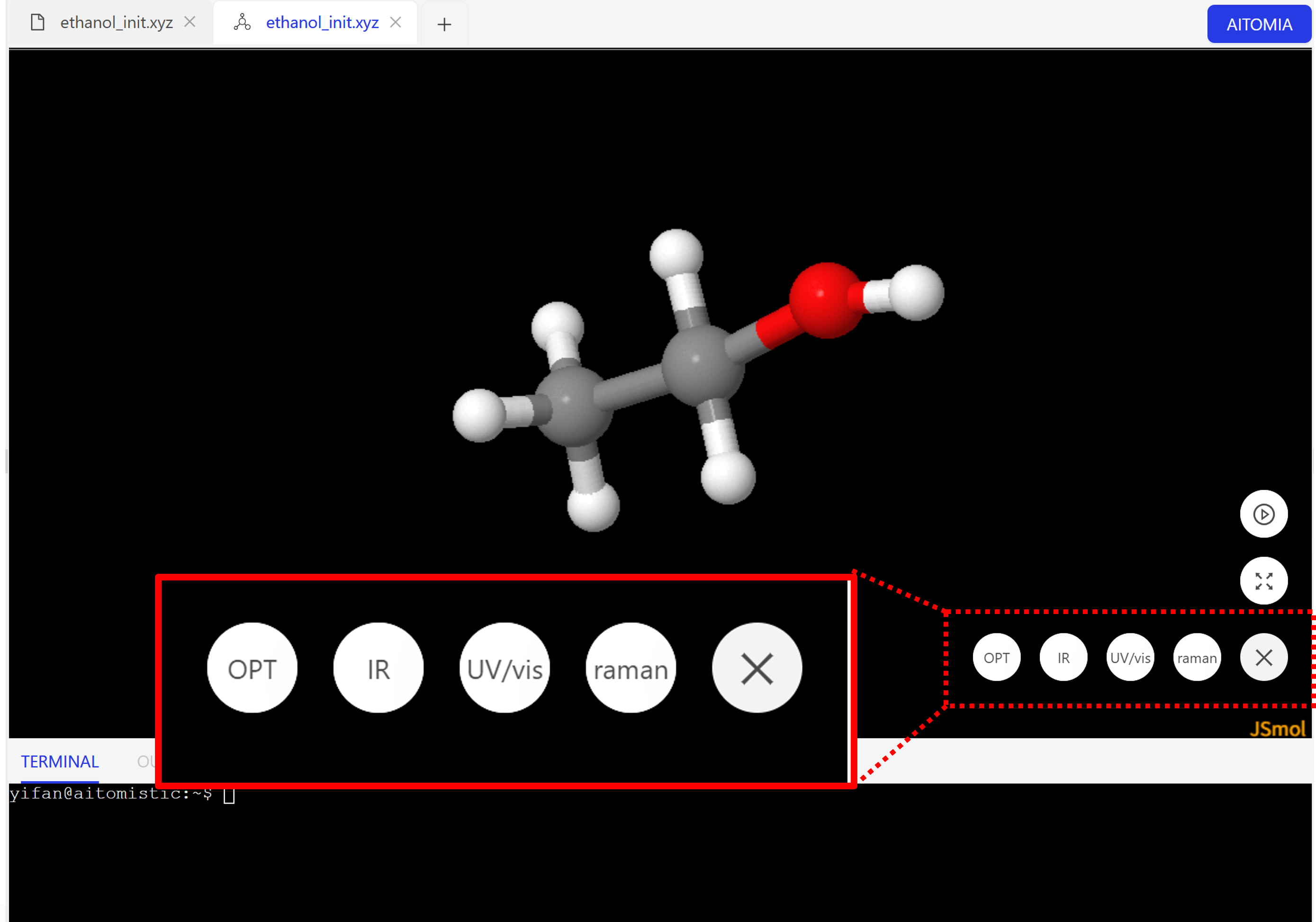
On Aitomistic Hub, you can easily generate the IR spectrum by simply clicking the button on the visualization page
Implicit solvent in AIQM2
Implicit solvent is applied by using keyword baseline_kwargs when defining the method. It can only be used via python script. The available solvents can be found here.
import mlatom as ml
aiqm2 = ml.model.methods(
method='AIQM2',
baseline_kwargs={'solvent':'water'})
Citations
If you have used AIQM2, then the following citations are appropriate in your publication:
Yuxinxin Chen, Pavlo O. Dral*. AIQM2: Organic Reaction Simulations Beyond DFT. Chem. Sci. 2025, accepted manuscript. DOI: 10.1039/D5SC02802G.
IR spectra with AIQM2: Yi-Fan Hou, Cheng Wang, Pavlo O. Dral*. Accurate and Affordable Simulation of Molecular Infrared Spectra with AIQM Models. J. Phys. Chem. A 2025, 129, 3613–3623. DOI: 10.1021/acs.jpca.5c00146.
MLatom: Pavlo O. Dral, Fuchun Ge, Yi-Fan Hou, Peikun Zheng, Yuxinxin Chen, Mario Barbatti, Olexandr Isayev, Cheng Wang, Bao-Xin Xue, Max Pinheiro Jr, Yuming Su, Yiheng Dai, Yangtao Chen, Lina Zhang, Shuang Zhang, Arif Ullah, Quanhao Zhang, Yanchi Ou. MLatom 3: A Platform for Machine Learning-enhanced Computational Chemistry Simulations and Workflows. J. Chem. Theory Comput. 2024, 20, 1193–1213. DOI: 10.1021/acs.jctc.3c01203.
MLatom: Pavlo O. Dral, Fuchun Ge, Yi-Fan Hou, Peikun Zheng, Yuxinxin Chen, Bao-Xin Xue, Max Pinheiro Jr, Yuming Su, Yiheng Dai, Yangtao Chen, Shuang Zhang, Lina Zhang, Arif Ullah, Quanhao Zhang, Yanchi Ou. MLatom: A Package for Atomistic Simulations with Machine Learning, version [add version number], Xiamen University, Xiamen, China, 2013–2024. MLatom.com.
GFN2-xTB* Hamiltonian: C. Bannwarth, S. Ehlert, S. Grimme, J. Chem. Theory Comput. 2019, 15, 1652–1671.
xtb program: Semiempirical extended tight-binding program package xtb.https://github.com/grimme-lab/xtb.
D4: E. Caldeweyher, C. Bannwarth, S. Grimme, J. Chem. Phys. 2017, 147, 034112.
D4 program: E. Caldeweyher, S. Ehlert, S. Grimme, DFT-D4, Version [check your version], (Mulliken Center for Theoretical Chemistry, University of Bonn, [year])
ANI model: J. S. Smith, O. Isayev, A. E. Roitberg, Chem. Sci. 2017, 8, 3192
TorchANI program: X. Gao, F. Ramezanghorbani, O. Isayev, J. S. Smith, A. E. Roitberg, J. Chem. Inf. Model. 2020, 60, 3408