Roaming, as an unusual and intriguing class of reaction, has attracted extensive attention, but its reaction pathway under external environmental interactions remains unclear. In a recent collaborative study carried out by Professor ZhiGang Wang (Jilin University) and Researcher HuaJie Song (Institute of Applied Physics and Computational Mathematics), they report for the first time the roaming process of nitrobenzene, which is influenced by the hydrogen bonds from water molecules. Remarkably, in the presence of these hydrogen bonds, the roaming reaction can still occur. The underlying mechanism responsible for the phenomenon is ascribed to the dramatically changed polarization and correlation interactions between the roaming radicals. Furthermore, the reaction rates and thermal perturbation probabilities are also remarkably influenced due to the presence of the hydrogen bonds, by approximately two orders of magnitude. It is hoped that this work will provide insights and guidance for understanding and modulating the intriguing roaming reaction. It is noted that the GKS-EDA calculations in this work were performed with XACS cloud computing.
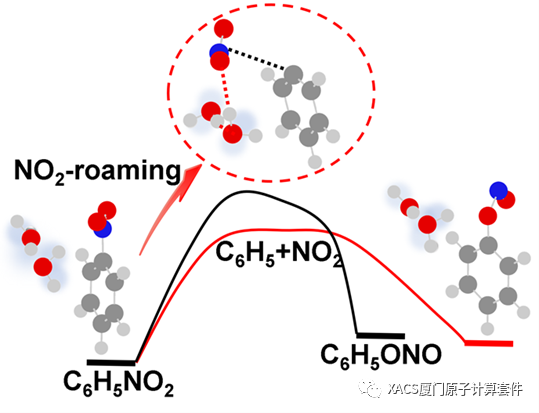
Through a search of the roaming transition state (TSR) and the verification of the intrinsic reaction coordinate method, the potential energy surfaces are obtained. The energy barriers of the roaming reactions for the isolation, the single and double water structures are 70.55 kcal/mol, 72.08 kcal/mol and 69.56 kcal/mol, respectively, indicating that the introduction of water molecules leads to a corresponding change in energy barrier. Despite barrier height is commonly employed for assessing chemical reactions, but the presence of a wider barrier in the double water structure could introduce certain uncertainties. These results confirm that the introduction of water molecules does not hinder the roaming reaction and underscores the need for further investigation.

Next, the GKS-EDA of the TSR structures was carried out to assess the influence of the H-bonds. The percentages of each attractive term suggests that the correlation and electrostatic terms were the most important for the radical-radical interaction, and the dispersion term was also important for the double water structure. In fact, the radical-radical interaction was strongly affected by the surrounding environment; thus, the many body effects, namely, ΔΔEint, require consideration as well. Their interaction energies for the single and double water structures were estimated as the sum of the radical-radical interaction energies and ΔΔEint. Due to the consideration of the many body effects, the correlation term became more dominant in the attractive terms, with its percentage significantly increasing from 40% for the isolation to 50% for the single water structure, respectively. Moreover, in repulsion interactions, the polarization term also significantly changed. In brief, these results indicated that the dramatically changed polarization and correlation interactions provided a possible electron correlation explanation for the influence of the H-bonds on the roaming reaction.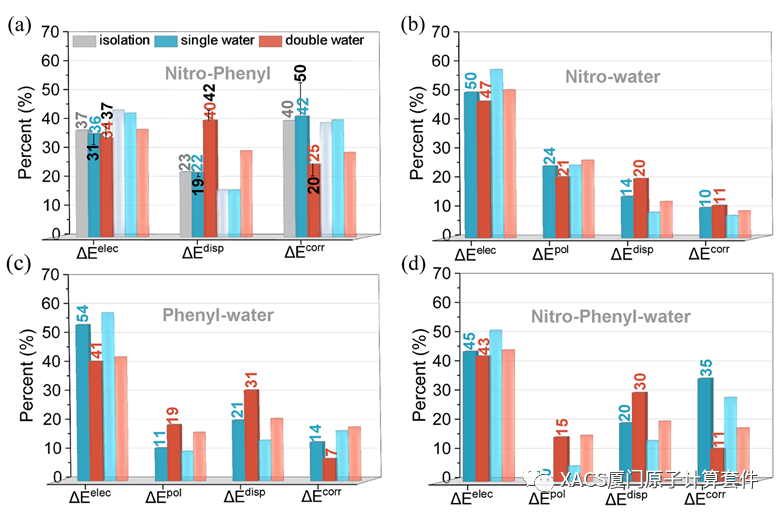
Radical-water interactions were also investigated. These interactions were primarily dominated by the electrostatic term. The electrostatic term of the radical-water interaction slightly decreased with the successive introduction of water molecules. As expected, the three-body nitro-phenyl-water interaction obtained from the wavefunction of the whole compounds was dominated by the electrostatic and correlation terms. Moreover, the polarization and dispersion terms were always small, indicating the weak covalent character of the radical-water interactions.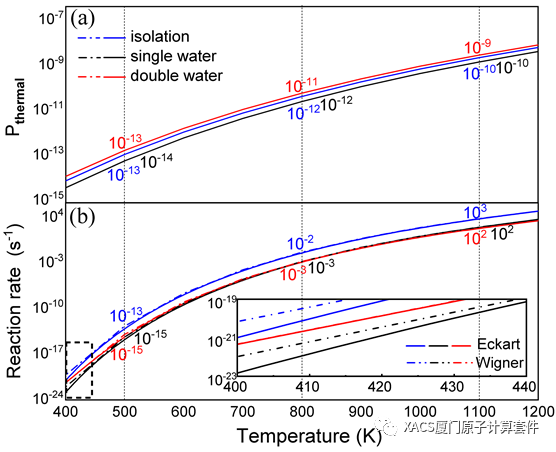
This work has provided a profound understanding of the roaming reaction under external environmental interactions, thereby inspiring the exploration of new approaches for controlling the roaming reaction.
Paper
Rui Liu #, Zhiyuan Zhang#, Longxiang Yan, Xinrui Yang, Yu Zhu, Peifeng Su*, Huajie Song*, and Zhigang Wang*. The Influence of Hydrogen Bonds on the Roaming Reaction. J. Phys. Chem. Lett. 2023, 14, 9351–9356. DOI: 10.1021/acs.jpclett.3c02133.