Tutorial on single-point calculations with MLatom@XACS
Published Time: 2024-02-14 09:29:51
This tutorial shows how to run single-point calculations with MLatom.
Single-point calculations can be performed with various models and methods in MLatom including:
Traditional QM methods
Hybird QM/ML methods
Pretrained-ML models and user-defined ML models
For more details on the models in MLatom, please check an overview. Here we will illustrate how to calculate isomerization energy of sugar in ISOL24 with different types of methods in MLatom.
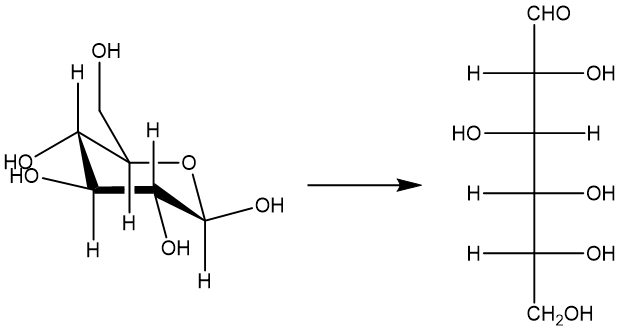
You can download the required file sugar_iso.xyz with geometries of two isomers in XYZ format (in Angstrom).
The basic idea here is that we will get single-point energies for isomers and calculate their energy difference. Since MLatom is a data-oriented package, it natively enables the calculations of many molecules. We first show how to run simulations via input files/command-line options and then how to get single-point properties in Python.
Single-point calculations with QM methods
MLatom supports single-point QM calculations with various popular programs such as Gaussian, PySCF, xTB, etc. Detailed manuals for simulations with QM methods can be found in Simulations with QM methods.
The typical input file for single-point calculations should contain xyzfile which defines geometry file and yestfile which defines the file to store energy.
For QM methods, users need to provide the method name and qmprog, i.e. the corresponding QM program.
Here is an example of the input file for energy-only calculations at B3LYP/6-31G* level with Gaussian
method=B3LYP/6-31G*
qmprog=gaussian
xyzfile=sugar_iso.xyz
yestfile=enest.dat
By running the command $mlatom mlatom.inp > mlatom.out, you will the get the MLatom output file as follows:
******************************************************************************
Energy of molecule 1: -687.1484077000000 Hartree
Energy of molecule 2: -687.1383648000000 Hartree
==============================================================================
Wall-clock time: 83.08 s (1.38 min, 0.02 hours)
MLatom terminated on 03.02.2024 at 09:35:57
==============================================================================
In the output file, energies of two molecules are listed in the same order as that in the input .xyz file.
Single-point calculations with hybrid QM/ML methods
One of the successful examples of hybrid QM/ML method is AIQM1, which is a general-purpose method approaching the gold-standard coupled cluster QM method at the cost of low-level semiempirical QM method. It takes the advantage of delta-learning strategy, correcting less accurate semiempirical method with state-of-art ML model ensemble (see figure below). Beyond this, in MLatom, users can also define their own delta-learning model, which is currently easy to achieve with Python API (see python API tutorial).
For AIQM1, users just need to request AIQM1 calculation in the first line:
AIQM1 # 1. request AIQM1 method
xyzfile=sugar_iso.xyz # 2. file with geometry
yestfile=enest.dat # 3. file to store energy
After running the command $mlatom mlatom.inp > mlatom.out, users will get the mlatom.out as :
******************************************************************************
Properties of molecule 1
Standard deviation of NN contribution : 0.00094895 Hartree 0.59548 kcal/mol
NN contribution : 0.00275644 Hartree
Sum of atomic self energies : -578.89384843 Hartree
ODM2* contribution : -107.70671483 Hartree
D4 contribution : -0.00358197 Hartree
Total energy : -686.60138879 Hartree
Properties of molecule 2
Standard deviation of NN contribution : 0.00066560 Hartree 0.41767 kcal/mol
NN contribution : 0.00159327 Hartree
Sum of atomic self energies : -578.89384843 Hartree
ODM2* contribution : -107.68954498 Hartree
D4 contribution : -0.00351388 Hartree
Total energy : -686.58531402 Hartree
==============================================================================
Wall-clock time: 0.77 s (0.01 min, 0.00 hours)
MLatom terminated on 03.02.2024 at 09:20:02
==============================================================================
For each molecule, all the energy components contained in AIQM1 method are presented, including ODM2* energy, D4 enengy and NN energy (sum of NN contribution and atomic self energies),
The standard deviation of NN contribution can be used to judge whether AIQM1 is reliable.
As a comparison, we show here isomeration energies calculated by each method and their respective times used.
Method |
\(\Delta E_\text{r}\) (kcal/mol) |
time (s) |
|---|---|---|
reference |
10.69 |
- |
AIQM1 |
10.08 |
0.77 |
B3LYP/6-31G* |
6.27 |
83 |
Single-point calculations with user-trained ML models
MLatom can read a user-trained model from a file and make predictions with it for new data (given either as input vectors X or as XYZ coordinates).
Users should provide either MLmodelType and/or MLprog argument. Below is the table for type of ML models and programs available:
MLmodelType |
MLporg |
|---|---|
KREG |
MLatomF |
sGDML |
sGDML |
GAP-SOAP |
GAP |
PhysNet |
PhysNet |
DeePot-SE |
DeepMD-kit |
ANI |
TorchANI |
MACE |
MACE |
We will use a pretrained MACE model to predict molecular enenrgy.
You can download mace model file here: mace.pt.
Here is an example of how to use user-trained MACE model to perform single-point calculation:
useMLmodel # 1. requests usage of ML model for prediction
MLmodelType=MACE # 2. request the MACE model
MLmodelIn=mace.pt # 3. the file with the trained model should be provided, here it is mace.pt file
xyzfile=sugar_iso.xyz # 4. file with geometries
For how to train ML models with MLatom, please refer to manual.
Gradients and Hessian
If users need gradients and Hessian, two lines can be added in the input file:
AIQM1 # 1. request AIQM1 method
xyzfile=sugar_iso.xyz # 2. file with geometry
yestfile=enest.dat # 3. file to store energy
ygradxyzestfile=grad.dat # 4. file to store gradients
hessianestfile=hess.dat # 5. file to store hessian
ygradxyzestfile will create the file storing the gradients and hessianestfile the Hessian. These keywords are case-insensitive but the file names are.
Charge and multiplicity
If molecules have other than default charges (default 0) and multiplicities (default 1), the input might look like:
method=B3LYP/6-31G*
qmprog=gaussian
xyzfile=sp.xyz
yestfile=enest.dat
charges=0,1
multiplicities=1,3
Calculations through PyAPI
Define methods
Python API provides a more flexible usage of MLatom methods. Detailed introduction is provided in method in python API.
For QM methods, AIQM1 and pre-trained ML models, user can define method by using mlatom.models.methods module.
There are three arguments here which are commonly used:
method: define the method you want to useprogram: the program to use the methodnthreads: number of threads used by the program
For example, you can define QM method as :
method = mlatom.models.methods(method='b3lyp/6-31G*', program='gaussian')
# method = mlatom.models.methods(method='b3lyp/6-31G*', program='pyscf', nthreads=18)
Note
AIQM1 use qm_program keyword to define the program used for ODM2* parts.
For user-trained ML models, they use different modules listed below. See manual of API for details on these modules.
model type |
module |
|---|---|
KREG |
|
sGDML |
|
GAP-SOAP |
|
PhysNet |
|
DeePot-SE |
|
ANI |
|
MACE |
|
Perform calculations
Here we will introduce the pipeline of using python API to calculate isomerization energy of sugar.
The energy unit used in MLatom is Hartree. Since the calculations are on multiple molecules, we start from using mlatom.data.molecular_database class to load these molecules and
use predict function in method to perform calculation
import mlatom as ml
# read molecule from .xyz file
molDB = ml.data.molecular_database.from_xyz_file('sugar_iso.xyz')
# define method
model = ml.models.methods(method='AIQM1')
# model = ml.models.methods(method='B3LYP/6-31G*'. program='pyscf') ## calculations using QM methods
model.predict(molecular_database=molDB)
print(molDB[0].energy)
print(molDB[1].energy)
print(molDB[1].energy-molDB[0].energy)
If gradients and hessian are needed, user can request gradient and hessian calculations by:
model.predict(molecular_database=mol, calculate_energy_gradients=True, calculate_hessian=True)
If only one molecules is used, user can also use mlatom.data.molecule class
mol = ml.data.molecule.from_xyz_file('sp.xyz')
and use it to perform the calculation:
model.predict(molecule=mol)
User can also assign charges and multiplicities to molecular_database or molecule by:
mol.charge = 1
mol.multiplicity = 3
molDB.charges = [1,1]
molDB.multiplicities = [1,3]
Retrieve properties
In MLatom, energy and Hessian will be stored in molecule object and gradients will be stored in atom object.
(Details in data in python API). User can request them by using
mol.energy
mol.get_energy_gradients()
mol.hessian
molecular_database provide funciton get_properties to extract properties from each molecule object.
You can retrieve an array of energy, gradients and hessians by using:
molDB.get_properties('energy')
molDB.get_xyz_vectorial_properties('energy_gradients')
molDB.get_properties('hessian')
Single-point calculations with user-defined hybrid models
MLatom possesses the power of combining different models together by use of mlatom.models.model_tree_node module, which works similar to tree structure
(details in model tree node in Python API)
Here we use a custom delta-learning model as an example, which was trained on the differences between full configuration interaction and Hartree–Fock energies of H2.
The pretrained model file can be downloaded here delta_FCI-HF_h2_ani.npz.
You can define 2 children nodes as:
baseline=ml.models.model_tree_node(name='baseline', operator='predict', model=ml.models.methods(method='HF/STO-3G', program='pyscf')
delta_correction=ml.models.model_tree_node(name='delta_correction', operator='predict', model=ml.models.ani(model_file='delta_FCI-HF_h2_ani.npz')`
You can see there is operator that operate on the model you defined (here each model just performs prediction task).
Now we define the parent node as:
ml.models.model_tree_node(name='hybrid', operator='sum', children=[baseline, delta_correction])
Here, the operator changes to sum since the operation we will perform on this node is summing up the predictions by two children nodes.
Finally we combine these things together and predict energy of H2. You can download the geometry file needed here h2_init.xyz.
import mlatom as ml
# Get the initial guess for the molecules to optimize
mol = ml.data.molecule.from_xyz_file('h2_init.xyz')
# Let's build our own delta-model
# First, define the baseline QM method, e.g.,
baseline = ml.models.model_tree_node(name='baseline', operator='predict', model=ml.models.methods(method='HF/STO-3G', program='pyscf'))
# Second, define the correcting ML model, in this case the KREG model trained on the differences between full CI and HF
delta_correction = ml.models.model_tree_node(name='delta_correction', operator='predict', model=ml.models.ani(model_file='delta_FCI-HF_h2_ani.npz'))
# Third, build the delta-model which would sum up the predictions by the baseline (HF) with the correction from ML
delta_model = ml.models.model_tree_node(name='delta_model', children=[baseline, delta_correction], operator='sum')
delta_model.predict(molecule=mol)
print(mol.energy)
Any questions or suggestions?
If you have further questions, criticism, and suggestions, we would be happy to receive them in English or Chinese via email, Slack (preferred), or WeChat (please send an email to request to add you to the XACS user support group).