Simulating ambimodal reactions online!
Published Time: 2024-07-10 20:53:00
Chemistry students learn early that a reaction proceeds from reactants to products via a transition state connecting them. This simple picture does not always hold though. The reactions never pass exactly through the transition state and we must propagate molecular dynamics trajectories to get a better view of what is actually happening. When we propagate such trajectories we may sometimes observe a curious phenomenon that the trajectories passing near the same transition state lead to different products. This is indeed a very well-documented phenomenon and such reactions are called ambimodal. They happen due to the post-transition state bifurcation of the potential energy surface.
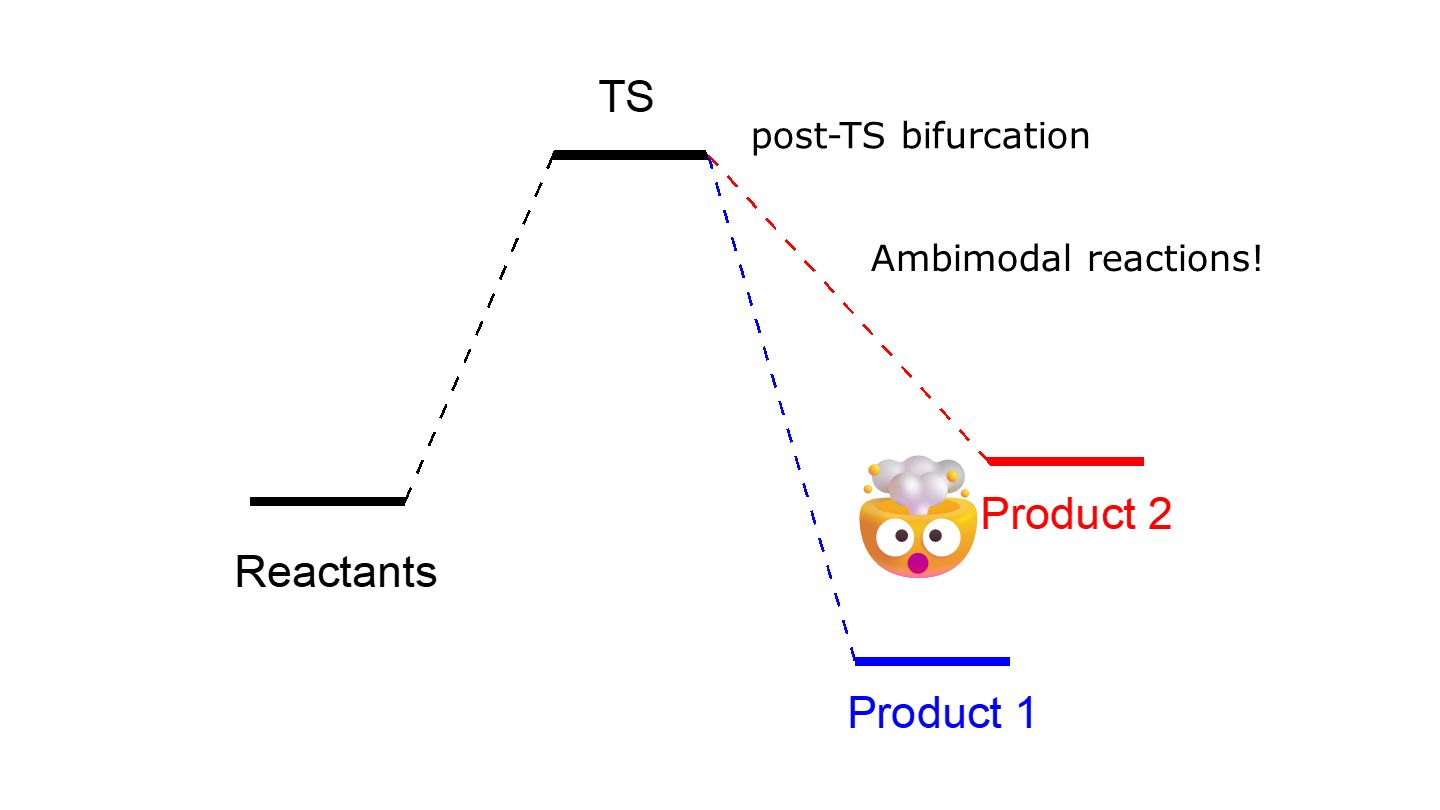
A popular approach to simulate the ambimodal reactions is to run downhill trajectories from the geometries sampled from real normal modes of the transition state – i.e., sampled orthogonally to the reaction coordinate. How to do that with quasi-classical trajectories, we’ve already covered before.
Here we will show how to perform simulations of ambimodal reactions routinely and accurately within one–two hours online!
We will
take this beautiful ambimodal [6+4]
cycloaddition as an
example.
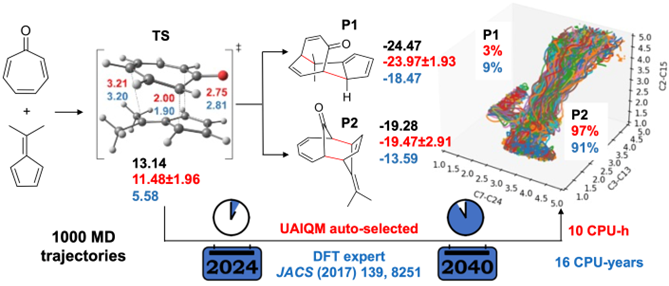
[Y. Chen, Y.-F. Hou, O. Isayev, P. O. Dral. Universal and Updatable Artificial Intelligence-Enhanced Quantum Chemical Foundational Models. 2024, submitted. https://doi.org/10.26434/chemrxiv-2024-604wb.]
Back in 2017, the state of the art was to perform 117 reactive trajectories with B3LYP. I can only imagine how much effort it took.
Now in 2024, we have at our disposal much faster and more accurate AI-enhanced quantum chemical methods: UAIQM which we introduced before. For this reaction, UAIQM provides accuracy better than B3LYP: just compare our UAIQM energies (in red) to the gold-standard coupled-cluster energies (in black). B3LYP energies in blue are quite off and that surely should impact the dynamics results. Indeed, our UAIQM calculations revise the product distribution from 10:1 at B3LYP to 30:1! Remarkably, you can now run the same simulations with UAIQM online in 1-2 hours and easily get these beautiful plots yourself with our script. You can use this script as a template for your reaction too.
Here we demonstrate how to do it.
First of all, go to our tutorial page. Go to our XACS cloud computing (if you are not registered – it is free). Then launch the Jupyter lab. We placed the tutorial scripts into jupyter_examples folder, in uaiqm subfolder (see the Jupyter notebook ambimodal.ipynb. To submit a job with on more CPU nodes for hundred of trajectories, please run sbatch ambimodal.sh). After you open it, you can run the script.
The script starts with loading mlatom as a Python library, then loading the initial guess of the transition state. Then the appropriate UAIQM model is chosen automatically for a given time budget for the single-point calculations (we set just 0.1 s as we want to get it fast!). Later, we use UAIQM to optimize the TS structure*, calculate frequencies and normal modes, do normal mode sampling, and finally launch a thousand trajectories in both forward and backward directions. We provide a sample routine to analyze the product ratio and plot them based on the key geometric parameters. You can also view beautiful trajectories.
*in script fewer trajectories are run for faster demonstration.
If you want to learn more about AI-enhanced computational chemistry, you can check out our new hands-on online mini-course, perfectly suitable for both beginners and experts wanting to upgrade their computational tools.