JCTC: Surface hopping dynamics with QM and ML methods
Published Time: 2024-06-12 20:58:15
XACS team in collaboration with Mario Barbatti and groups in Warsaw University and Zhejiang lab has recently published a paper in JCTC about the versatile Python implementation of surface-hopping dynamics.
This implementation is based on a powerful MLatom ecosystem for AI-enhanced computational chemistry. Using Python script templates with just dozens of lines, the users can perform the full computational workflow from geometry optimization to normal mode sampling to surface hopping dynamics and analysis.
The user has a rich choice of ab initio and semi-empirical quantum chemical and machine learning methods for performing the dynamics.
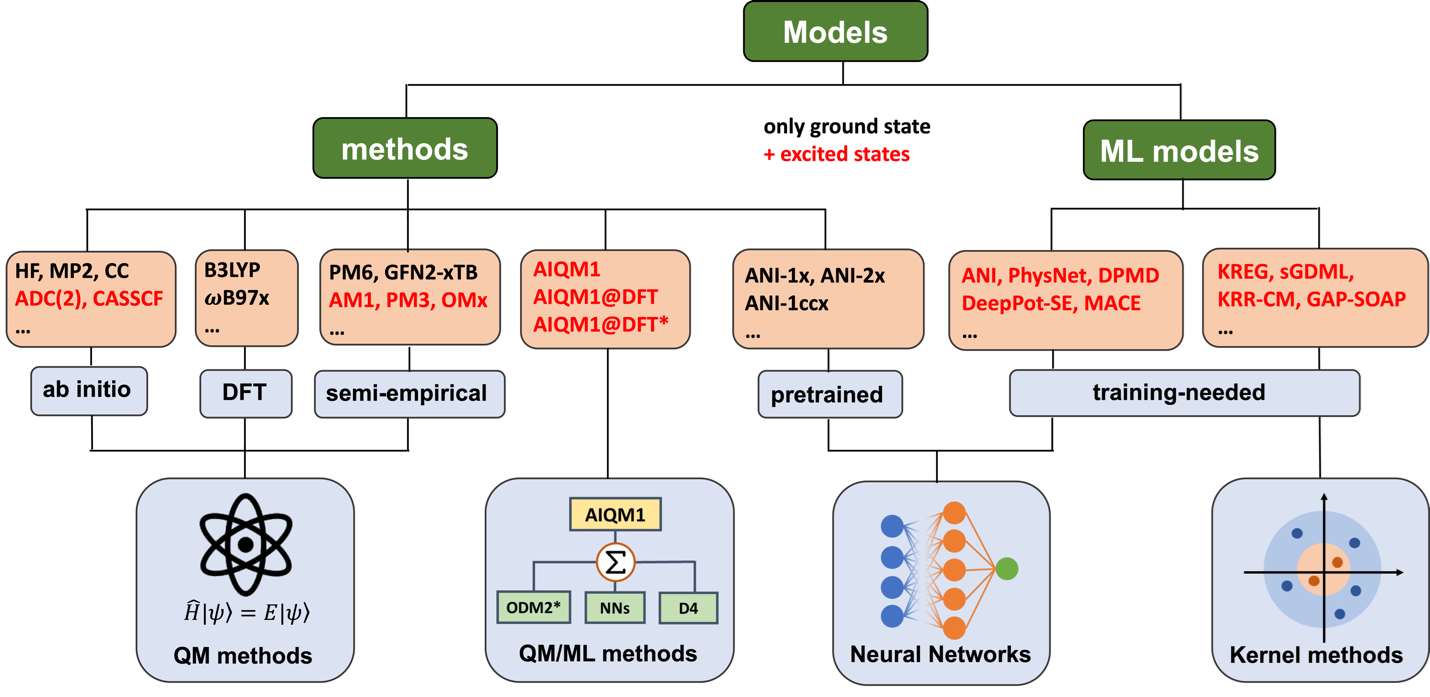
[Figure
credit: Modified from P. O. Dral, F. Ge, Y.-F. Hou, P. Zheng, Y. Chen, M.
Barbatti, O. Isayev, C. Wang, B.-X. Xue, M. Pinheiro Jr, Y. Su, Y. Dai, Y.
Chen, S. Zhang, L. Zhang, A. Ullah, Q. Zhang, Y. Ou. JCTC 2024, 20,
1193 under CC-BY license]
Among them, ML-based AIQM1 may deliver rather good surface hopping dynamics with a reduced computational cost.
For more details on how to setup the calculations, you can check our online tutorials and the online broadcast where you can listen to an expert discussion on the implementation and the demonstration of the machine learning surface hopping dynamics performed within seconds on our online XACS platform.