Test Calculation
VBSCF calculation of HF molecule
precise integrals
HF molecule, 3 structures
$ctrl
vbscf # request VBSCF computation
str=full nao=2 nae=2 # automatically generate all 3 structures
orbtyp=hao frgtyp=sao
int=libcint # precise integral generated by libcint
basis=3-21G
$end
$frag
1 1 1 1
s 1
spz 2
px 2
py 2
$end
$orb
1 1 1 1 1 1
2
2
3
4
1
2
$end
$geo
H 0.0 0.0 0.0
F 0.0 0.0 0.9
$end
Tip
VB structures are generated automatically by ,
STR=FULL NAO=2 NAE=2so$STRis not neededVB orbitals are described with
SAO. See the$FRAGand$ORB.
VBSCF with RI
HF molecule, 3 structures
$ctrl
vbscf # request VBSCF computation
str=full nao=2 nae=2 # automatically generate all 3 structures
orbtyp=hao frgtyp=sao
int=ri # integral evaluated by RI
basis=3-21G
$end
$frag
1 1 1 1
s 1
spz 2
px 2
py 2
$end
$orb
1 1 1 1 1 1
2
2
3
4
1
2
$end
$geo
H 0.0 0.0 0.0
F 0.0 0.0 0.9
$end
BOVB calculation with RI
This example shows the BOVB computation for benzylacetamide dimer. The number of basis function is 550.
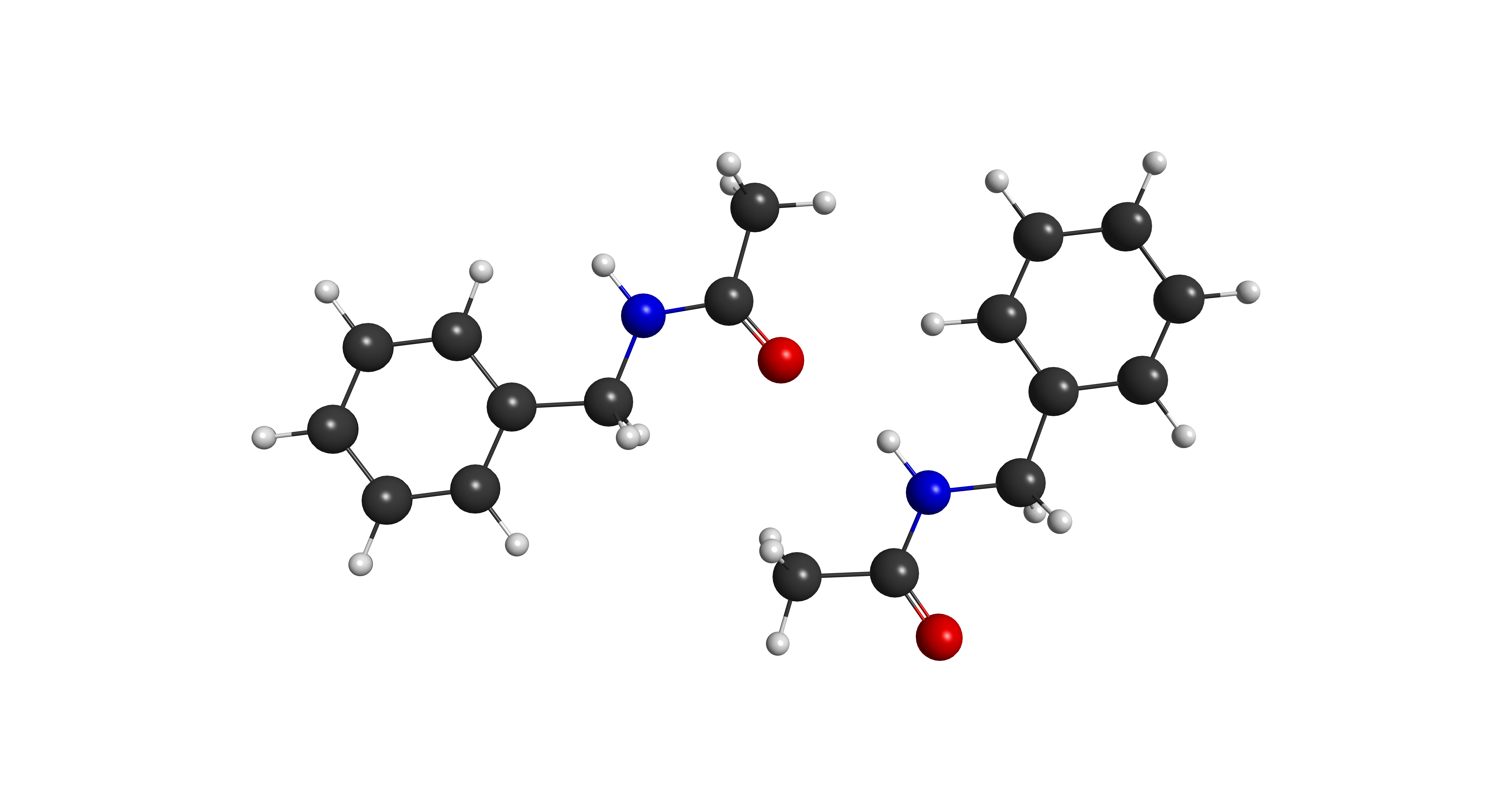
The user may download the input file, try the computation themselves and compare with the output file.
VBSCF calculation with COSX
This exmaple shows the VBSCF computation of piperdine-C60 with COSX. Number of basis function in this computation is 1012.
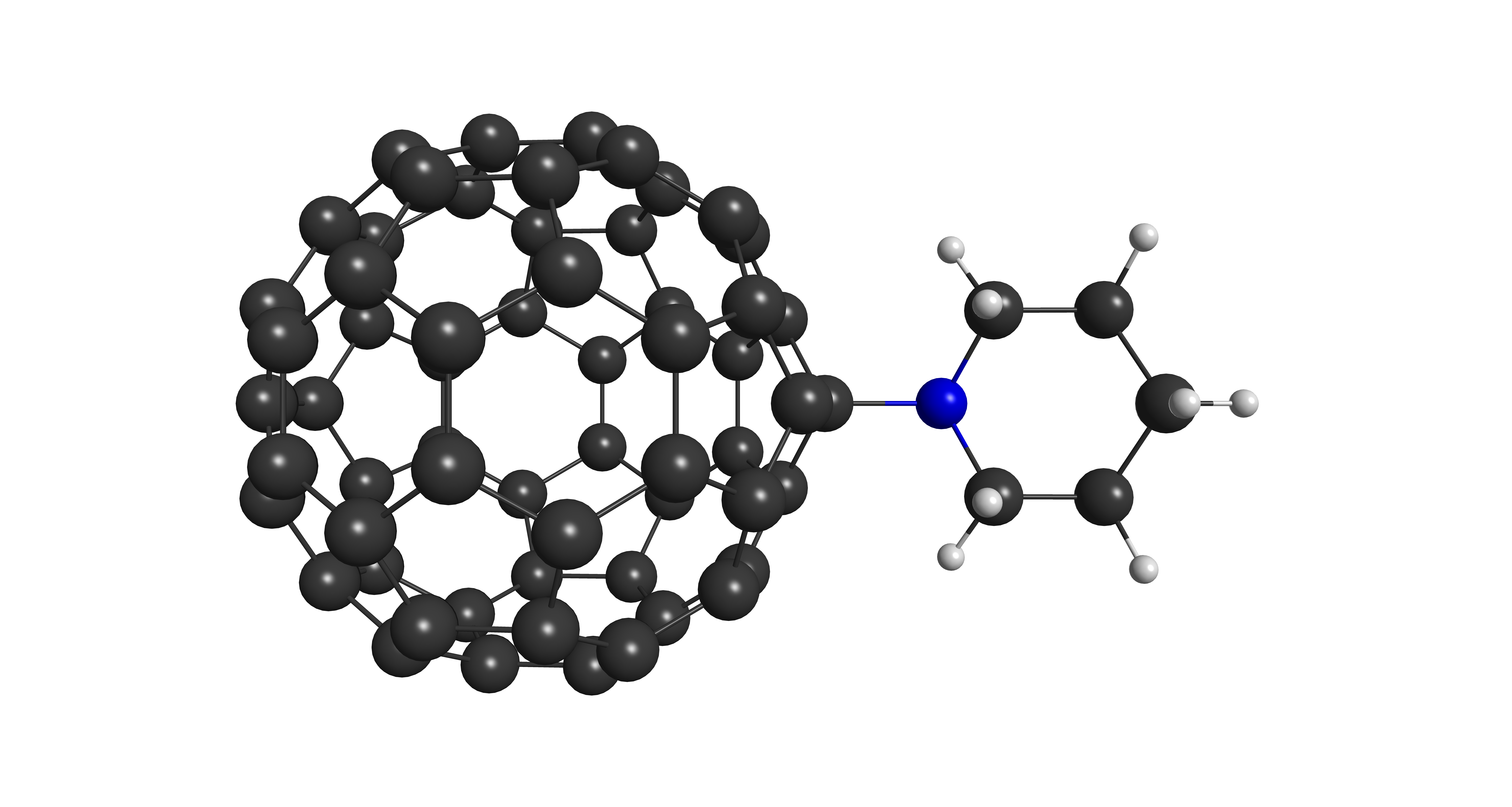
The user may download the input file and try the computation themselves.
\(\lambda\)-DFVB(U) Calculation of the TS of an \(SN\)2 reaction
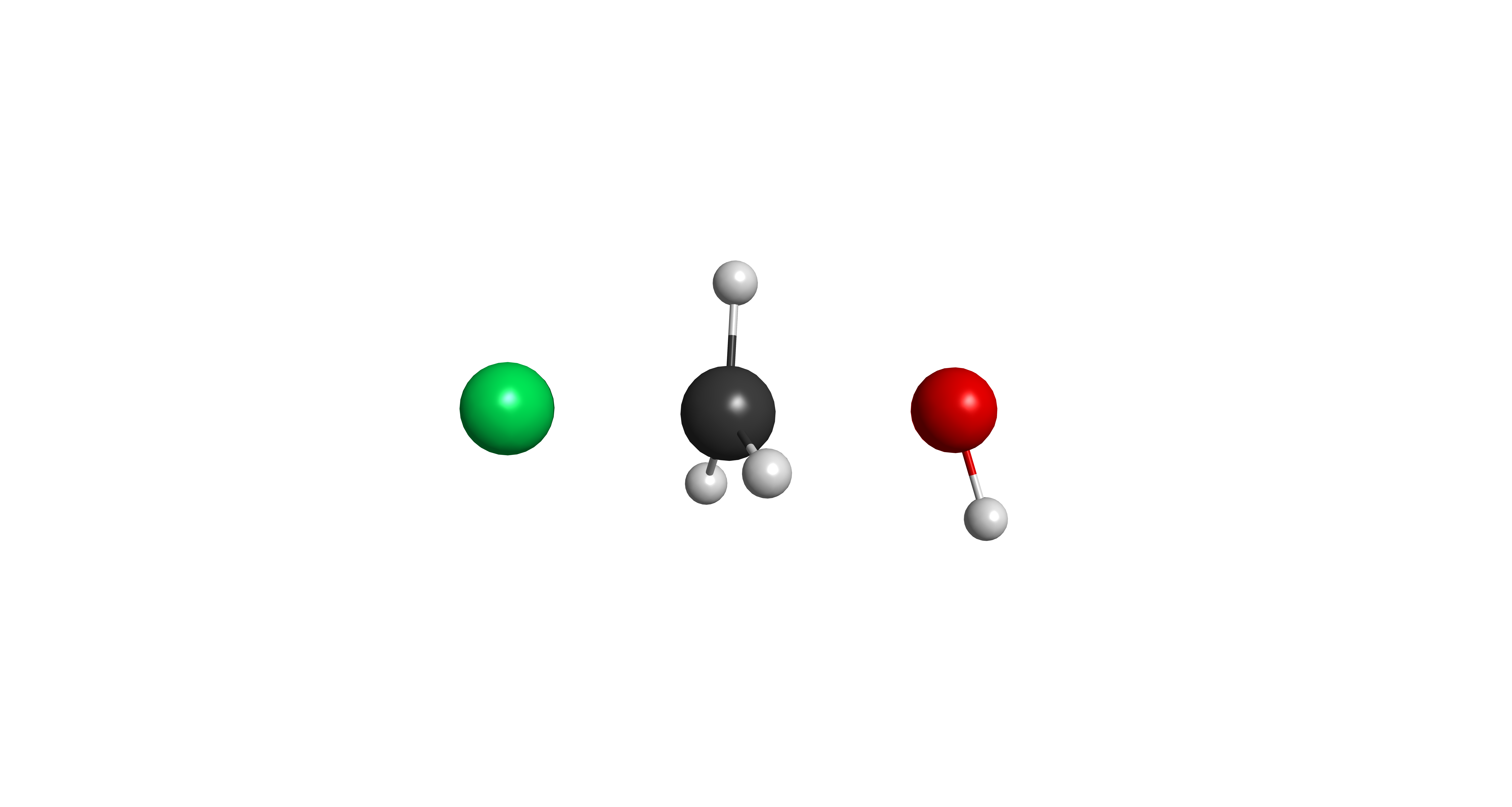
The user may download the input file and try the computation.
Tip
The inisial guess is the wave function with
STR=FULLandHAO. The orbitals in the initial guess are localized on specific atoms or bonds.
λ-DFVB(MS) Calculation of the Spiro cation
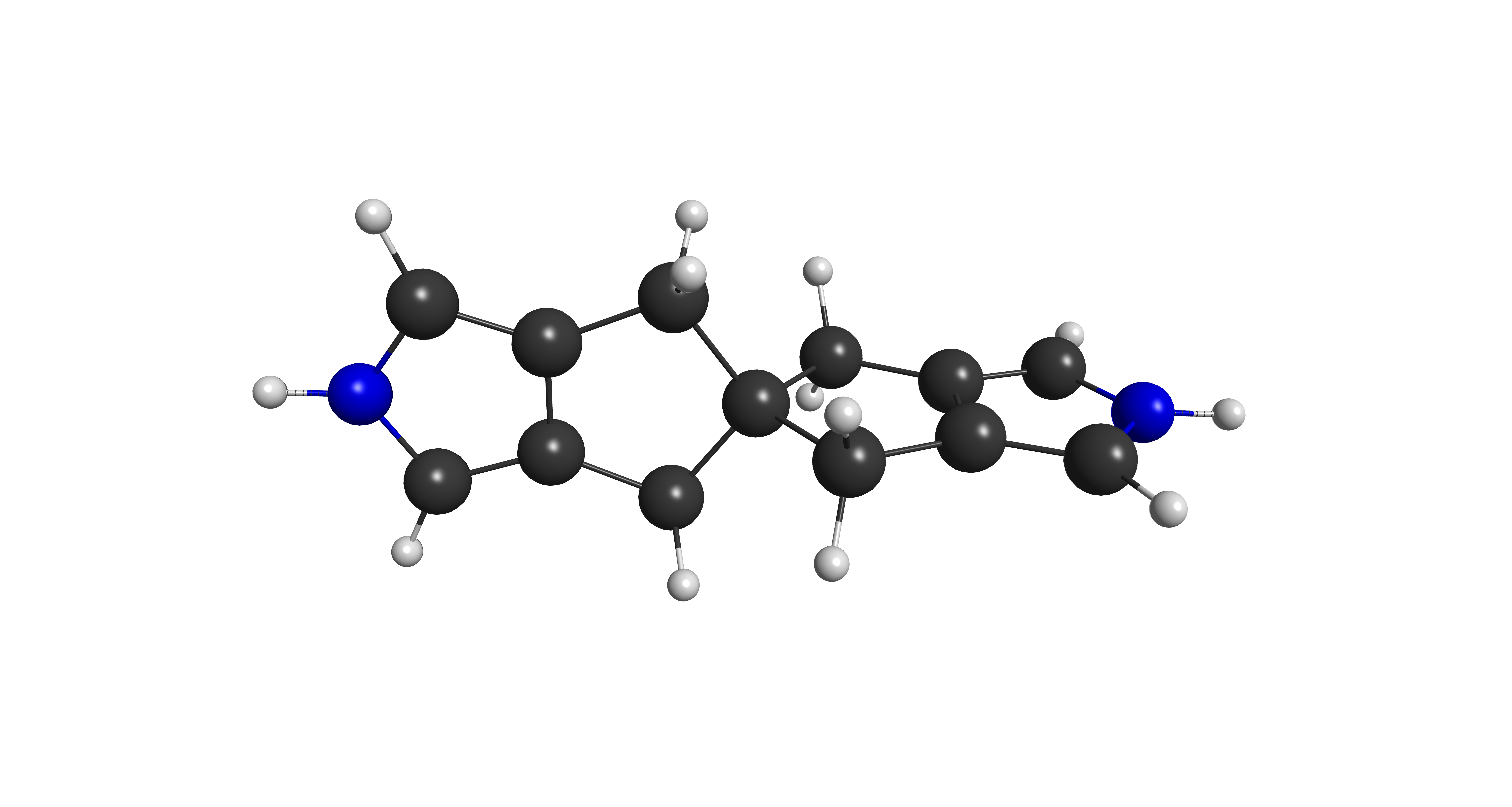
The input file is attached here.
VBCAD Analysis of LiF Molecule
LiF VBCAD
$ctrl
vbscf
str=full nae=2 nao=2
iscf=5
int=libcint basis=aug-cc-pvtz
wstate(1)=1,1 vbcad itmax=500
$end
$actorb
1
2
$end
$geo
F 0.0 0.0 0.0
Li 0.0 0.0 3.8
$end
$gus
1 1
2 2
3 3
4 4
5 5
6 6
7 7
$end
Tip
By using the simplified input format, the
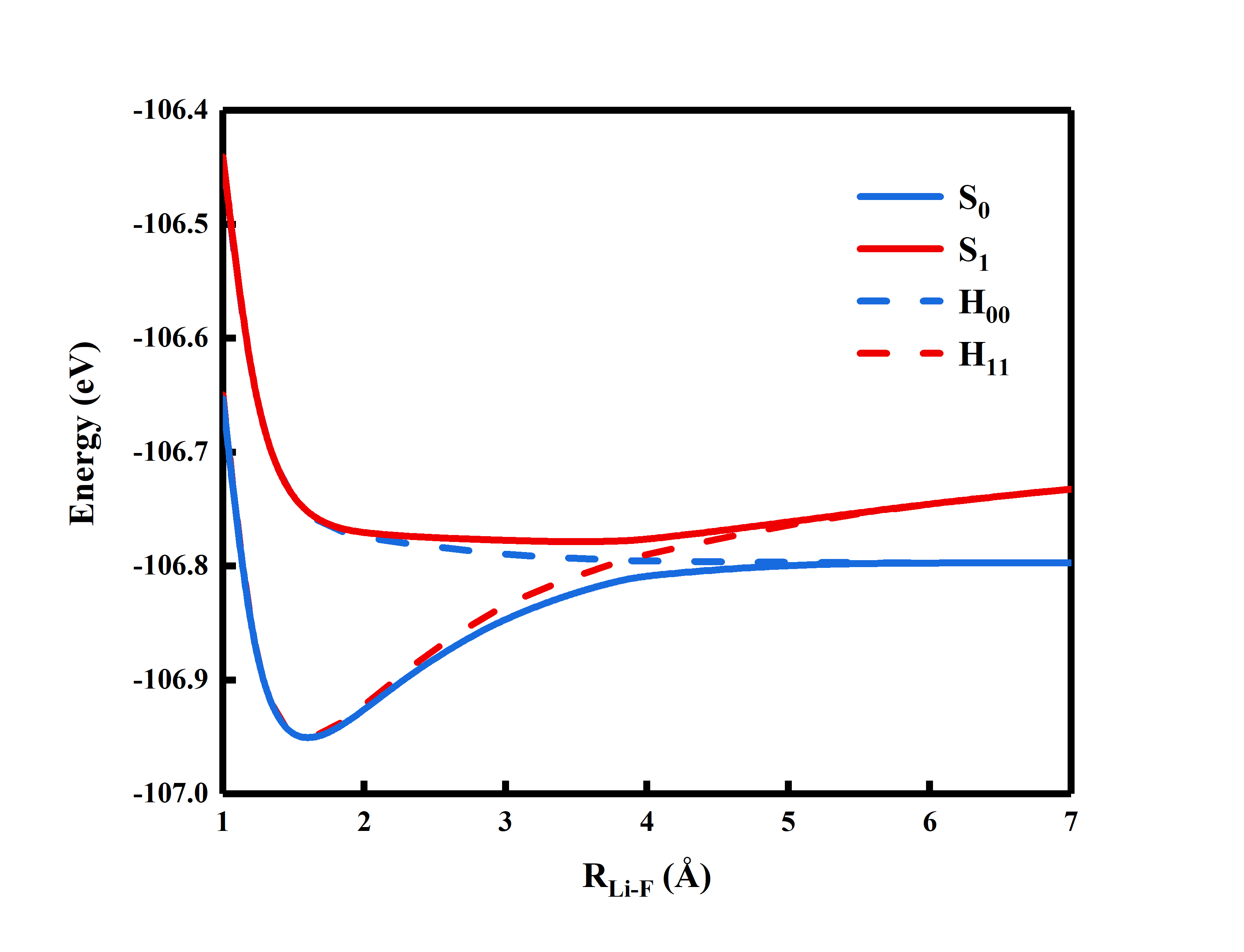
$ACTORBsection only required user to input the active orbitals.Users are encouraged to experiment with different bond lengths at RLi-F = 1.0, 1.2, 1.4, 1.6, 1,8, 2.0, 2.4, 2.6, 2.8, 3.0, 4.0, 7.0 Angstrom, get the energies and plot the potential energy surface. See at which distance the diabatic states cross. Following is the example of the potential energy surface.
To scan the potential energy surface, it is recommended that the user replace the content of
$GUSby the content of.orbfrom the points nearby.
Geometry Optimization of the Kekulé structure of benzene molecule
Kekule benzene Gopt
$ctrl
vbscf
nstr=1 nae=6 nao=6
iscf=5
int=libcint basis=cc-pvdz
itmax=500
opt
$end
$str
1:18 19 20 21 22 23 24
$end
$actorb
1
2
3
4
5
6
$end
$geo
C 0.000000 1.395000 0.000000
C 1.208000 0.697000 0.000000
C 1.208000 -0.697000 0.000000
C 0.000000 -1.395000 0.000000
C -1.208000 -0.697000 0.000000
C -1.208000 0.697000 0.000000
H 0.000000 2.495000 0.000000
H 2.161000 1.247000 0.000000
H 2.161000 -1.247000 0.000000
H 0.000000 -2.495000 0.000000
H -2.160000 -1.247000 0.000000
H -2.160000 1.247000 0.000000
$end
$gus
1 1
2 2
3 3
4 4
5 5
6 6
7 7
8 8
9 9
10 10
11 11
12 12
13 13
14 14
15 15
16 16
17 18
18 19
19 17
20 17
21 17
22 17
23 17
24 17
$end
Geometry Optimization of the Dewar structure of benzene molecule
Dewar benzene Gopt
$ctrl
vbscf
nstr=1 nae=6 nao=6
iscf=5
int=libcint basis=cc-pvdz
itmax=500
opt
$end
$str
1:18 19 20 21 24 22 23
$end
$actorb
1
2
3
4
5
6
$end
$geo
C 0.000000 1.395000 0.000000
C 1.208000 0.697000 0.000000
C 1.208000 -0.697000 0.000000
C 0.000000 -1.395000 0.000000
C -1.208000 -0.697000 0.000000
C -1.208000 0.697000 0.000000
H 0.000000 2.495000 0.000000
H 2.161000 1.247000 0.000000
H 2.161000 -1.247000 0.000000
H 0.000000 -2.495000 0.000000
H -2.160000 -1.247000 0.000000
H -2.160000 1.247000 0.000000
$end
$gus
1 1
2 2
3 3
4 4
5 5
6 6
7 7
8 8
9 9
10 10
11 11
12 12
13 13
14 14
15 15
16 16
17 18
18 19
19 17
20 17
21 17
22 17
23 17
24 17
$end