6. Spectroscopy
This tutorial will use MLatom@XACS program. See its manual and tutorials for more details.
Here we will show how to use MLatom for different spectroscopic simulations:
6.1. Infrared spectra
In this example, we generate infrared (IR) spectrum of ethanol from an existing MD trajectory propagated with AIQM1 for 3 ps. See previous section on how to run dynamics. AIQM1 also generates dipole moments which are needed to calculate intensities in infrared spectra.
6.1.1. Input file
MLatom input is very simple:
IRSS # Infrared Spectra Simulation
TrajH5MDIn=traj.h5 # H5MD file
where the file traj.h5 is pre-acquired trajectory file with information of dipole moments inside.
6.1.2. Computational results
You can see the spectrum (ir.png) in your folder after the computation is finished:
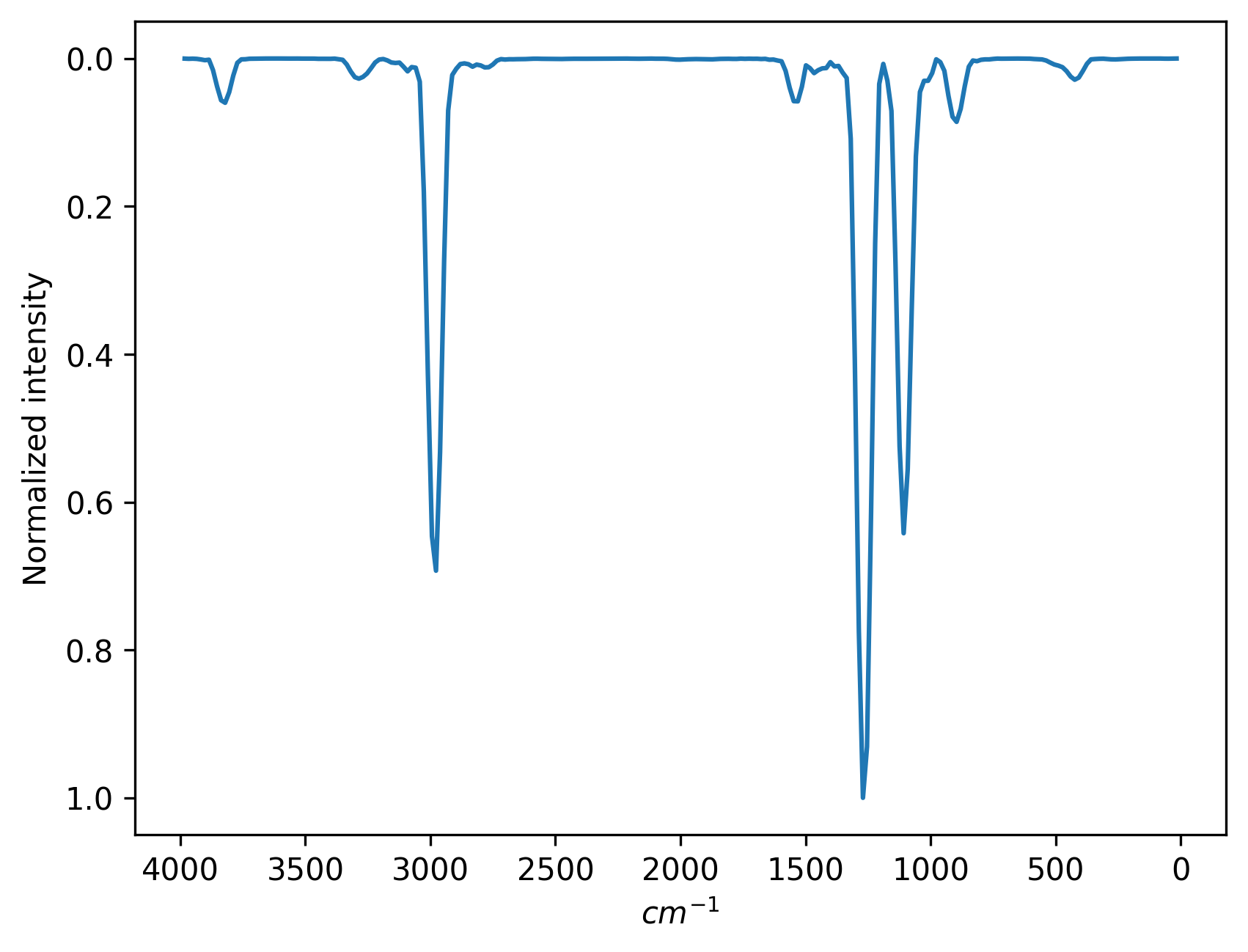
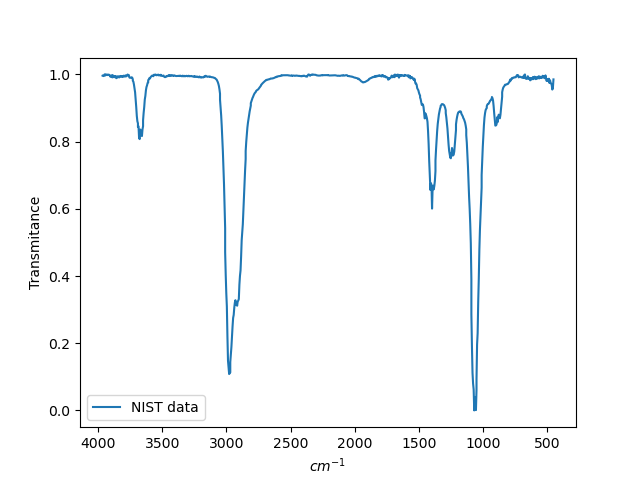
Below, for comparison, is the spectrum from the NIST database:
6.2. UV/vis absorption spectra with machine learning
Machine learning can be used to greatly accelerate the calculation of precise UV/vis absorption spectra via nuclear ensemble approach (NEA) which typically requires many hundreds or thousands of expensive quantum mechanical calculations of excited states. MLatom supports such ML-accelerated NEA calculations as described in our paper.
The following example is from our book chapter.
6.2.1. Input files
MLatom input file:
cross-section
Nexcitations=30
plotQCNEA
plotQCSPC
deltaQCNEA=0.05
These calculations require many data files (reference excitation energies at TDDFT level).
These data files are zipped and should be uploaded as a zip archive to the cloud computing as auxiliary file.
6.2.2. Computational results
Calculations can take more than 5 min. MLatom automatically determines the minimum required number of training points, in this case it needed 200 points for precise spectrum. In the output file you can find that it took 4 iterations to converge:
==========================================================================================
run ML-NEA iteratively for spectrum generation ( ML_train_iter ) started at Wed Dec 1 12:00:19 2021 CST
ML-NEA iteration 1: train_number = 50; RMSE_geom = 0.06717941145022376; rRMSE = 1.0
ML-NEA iteration 2: train_number = 100; RMSE_geom = 0.09043318436728051; rRMSE = 0.25713761026721255
ML-NEA iteration 3: train_number = 150; RMSE_geom = 0.06411060145373663; rRMSE = 0.410580813729204
ML-NEA iteration 4: train_number = 200; RMSE_geom = 0.0695737045717655; rRMSE = 0.07852252732055763
ML-NEA iteration ended after 4 iteration!
run ML-NEA iteratively for spectrum generation ( ML_train_iter ) finished at Wed Dec 1 12:08:01 2021 CST |||| total spent 462.02 sec
==========================================================================================
After the calculations finished, the spectra are plotted to plot.png file in the cross-section sub-directory. It should look like:
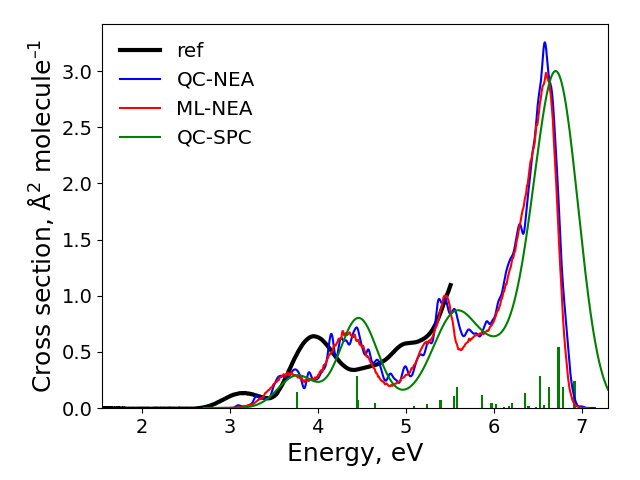
The final result: ‘ref’ is the experimental spectrum, QC-NEA – spectrum calculated with quantum chemical approach on 200 points in ensemble, ML-NEA – machine learning spectrum generated with 200 points in the training set and 50k points in ensemble, QC-SPC – spectrum generated with single-point convolution.
6.2.3. Questions
Compare obtained spectra and answer the following questions:
What are the problems with the QC-NEA spectrum?
What are the problems with the QC-SPC spectrum?
How to improve accuracy of the ML-NEA spectrum?
6.3. Two-photon absorption cross sections with machine learning
Two-photon absorption (TPA) is an important physical phenomenon which can be exploited in many different applications like upconverted laser. MLatom implements machine learning method predicting TPA cross section for a new molecule just by providing its SMILES (see this paper in Adv. Sci. for details).
6.3.1. Input files
Here we show how to calculate TPA cross section for RHODAMINE 6G and RHODAMINE 123 molecules with MLatom input file mltpa.inp:
MLTPA
SMILESfile=Smiles.csv
auxfile=_aux.txt
This input requires Smiles.csv file with SMILES of molecules:
CCNC1=CC2=C(C=C1C)C(=C3C=C(C(=[NH+]CC)C=C3O2)C)C4=CC=CC=C4C(=O)OCC.[Cl-]
COC(=O)C1=CC=CC=C1C2=C3C=CC(=N)C=C3OC4=C2C=CC(=C4)N.Cl
and optional _aux.txt, which defines the wavelength_lowbound, wavelength_upbound, and Et30 for making predicitons:
600,850,55.4
600,600,33.9
After you prepared your input files mltpa.inp, Smiles.csv, and _aux.txt, you can run MLatom as usual.
6.3.2. Computational results
After the calculations finish, the predicted TPA cross section values are saved in two files for two molecules: tpa1.txt and tpa2.txt. For our examples, they look like:
wavelength,predicted_sigma (GM)
600.0,285.19455
610.0,297.71707
620.0,284.11694
......
810.0,121.51988
820.0,116.537994
830.0,118.04909
840.0,103.65925
850.0,113.72374
and
wavelength,predicted_sigma (GM)
600.0,138.2346