Data
Here we show how to use MLatom to deal with data. There are three major data types in MLatom: molecules, databases of molecules, and MD trajectories, which are shown below (figure from MLatom 3 paper under CC-BY license):
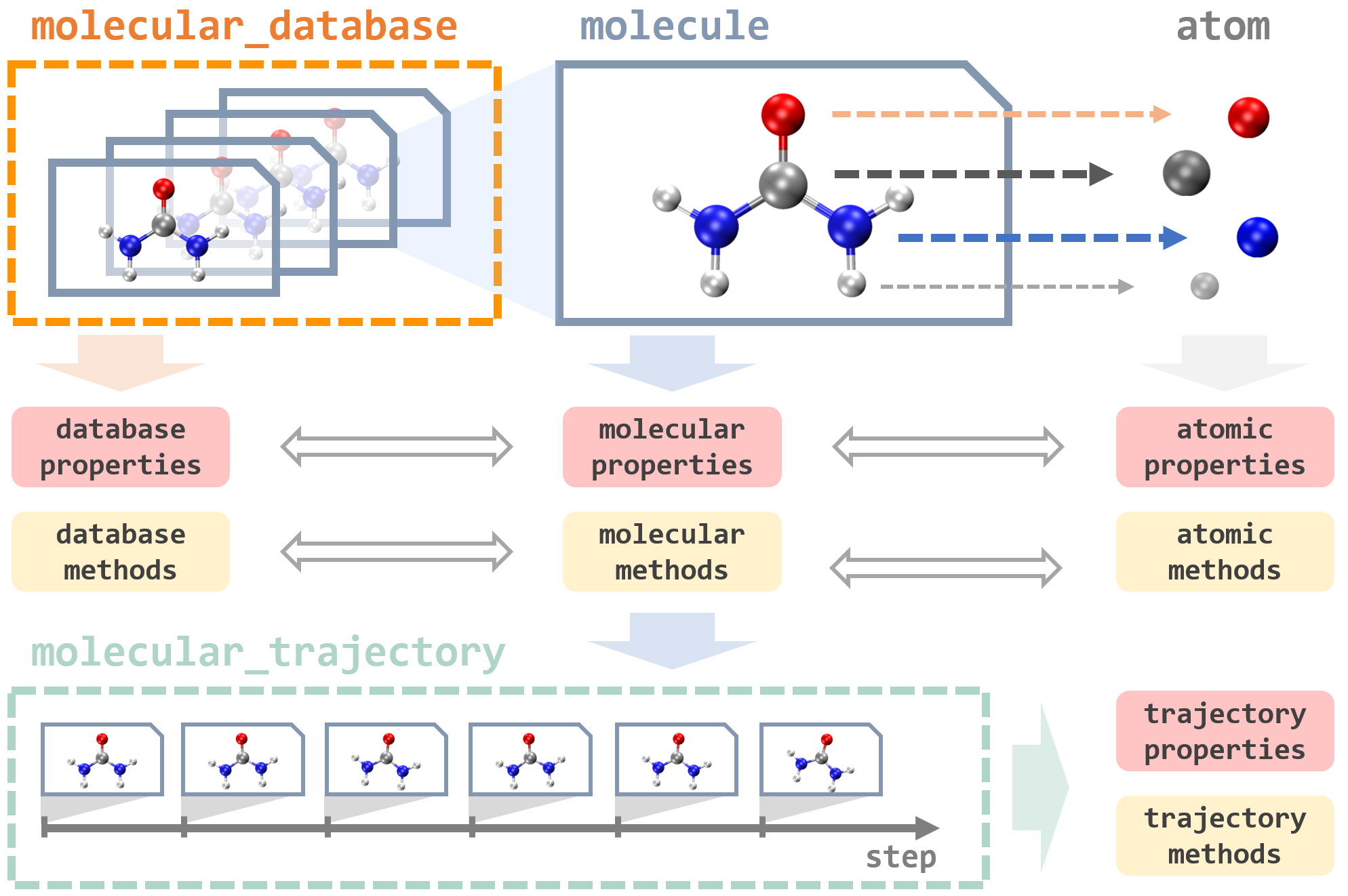
Before running examples below in Python/Jupyter notebook, you need to add and run the line import mlatom as ml as usual. We also provide a short video introduction about data objects used in MLatom:
Molecules
Molecule is a central concept in MLatom as most of operations are usually done on molecule class objects. This objects store the information about the constituents atom objects, their coordinates, and any property we want to learn or calculate. Molecules can be loaded and dumped in various formats.
Creating and loading molecule
A molecule object can be created from scratch. A very explicit way (not used in practice!) relecting how the molecule is actually constructed is the following:
mol = ml.molecule()
atom1 = ml.data.atom(atomic_number=1, xyz_coordinates=[0.0, 0.0, 0.0])
mol.atoms.append(atom1)
...
In practice, you can create the molecule using many useful functions. E.g., you can load it from SMILES string and the geometry will be generated and optimized with Pybel’s make3D() method:
mol = ml.molecule()
mol.read_from_smiles_string('CCCCCCC')
print(mol.xyz_coordinates)
...
In above example, you can see how to access the XYZ coordinates stored in a molecule object.
Very common way is to load the geometry from XYZ coordinates. There are several ways to do it. You can either provide XYZ coordinates as a string, read them from file or from numpy array:
mol = ml.molecule()
mol.read_from_xyz_string('''2
H 0 0 0
H 0 0 0.74
''')
# or
mol.read_from_xyz_file('h2.xyz')
# or
import numpy as np
xyz = np.array([[0, 0, 0], [0, 0, 0.7]])
mol.read_from_numpy(coordinates=xyz, species=np.array([1, 1]))
...
A small nuance is that if you remove read_ from above methods, you can directly get the molecule object, i.e., compare:
mol = ml.molecule()
mol.read_from_xyz_file('h2.xyz')
# or
mol = ml.molecule.from_xyz_file('h2.xyz')
# also works:
mol = ml.molecule.from_smiles_string('CCCCCCC')
mol = ml.molecule.from_numpy(coordinates=xyz, species=np.array([1, 1]))
Also, you can read XYZ files in different formats. The default one is:
first line: number of atoms
second line: blank or comments
following lines: element symbol or atomic number followed by x, y, z coordinates in Angstrom
MLatom also supports reading XYZ files in Columbus, Newton-X, and Turbomole formats, just add format, e.g., as mol = ml.molecule.from_xyz_file('h2.xyz', format='Newton-X').
Dumping and loading
Ultimately, molecule is a data format. Hence, it is convenient to dump and load it as such, e.g., in json format:
mol.dump(filename='h2.json')
mol2.load(filename='h2.json')
print(mol, mol2)
You can also save the coordinates in text file:
mol.write_file_with_xyz_coordinates('h2.xyz')
or load the molecule from Gaussian output file and dump it later:
mol = ml.molecule.load('gaussian_output_file.log', format='gaussian')
mol.dump('parsed_molecule.json', format='json')
Properties
You can access, modify, and add different properties.
Some of the most common examples are charge and multiplicity:
mol.charge = 1
mol.multiplicity = 2
We have already saw how to access xyz coordinates. In the same way, we can access other properties (if they are availble, of course…), e.g.:
# examples of accessing properties
xyz = mol.xyz_coordinates
print(mol.energy)
print(mol.energy_gradients)
mol.hessian
# modifying properties
mol.energy = 3.00
mol.energy_gradients = np.array(...)
A bit more complicated are properties corresponding to different electronic states (see a dedicated tutorial on excited-state data format). They are stored in molecule.electronic_states which is a list of molecule objects themselves, corresponding to each state (note that by default this list is empty – it is only filled either after the excited-state calculations or by the user). This allows to access a property of any state the same way as accessing a property of a ‘normal’ molecule object. For example:
# let's get S1 state
S1 = mol.electronic_states[1]
print(f'Energy of S1 state is {S1.energy}')
print(f'Its forces are')
print(-S1.energy_gradients)
print(f'checking multiplicity: {S1.multiplicity}')
Other useful methods
Above overview is just to explain the key features when dealing with molcules. There is much more to it, which you can check out in the manual:
getting xyz string for printing it
calculating and rescaling kinetic energy
getting internuclear distances (for a pair and all pairs of atoms)
copying molecules with or without indicated properties
getting smiles (
molecule.smiles)rotating, translating, scaling, and aligning
printing detailed info (with
molecule.info())getting numpy array with electronic state energies, gradients, energy gaps, and excitation energies
dealing with generic vectorial properties (e.g., dipole moments)
and some others more technical methods.
Molecular databases
We often have to deal with a collection of molecular objects, which are stored in the
molecular_database class instances. The molecules are stored in the list molecular_database.molecules.
Molecular databases are in many respects extension of the molecule class, e.g., they can be loaded and dumped in a similar manner:
# prepare H2 geometries with bond lengths ranging from 0.5 to 5.0 Å
xyz = np.zeros((451, 2, 3))
xyz[:, 1, 2] = np.arange(0.5, 5.01, 0.01)
z = np.ones((451, 2)).astype(int)
molDB = ml.molecular_database.from_numpy(coordinates=xyz, species=z)
molDB.dump(filename='h2.json')
molDB2 = ml.molecular_database.load(filename='h2.json')
The useful feature of molecular databases is that they can be manipulated as lists/numpy arrays:
print(len(molDB)) # 451
print(len(molDB[:100:4])) # 25
A related feature is that a database can be split into several other databases (useful, e.g., for splitting into training and test sets):
# let's change the splitting to 9:1 instead of default 8:2
train, test = molDB.split(fraction_of_points_in_splits=[0.9, 0.1])
Molecular databases also provide an easy acccess to the properties of all molecules, e.g., if we want to get the list of some property and save them in a text file, we can use:
molDB.get_properties(property_name='energy')
molDB.write_file_with_properties('h2.energies', property_name='energy')
There are also options to dump gradients and other vectorial properties as well as other useful technical features, see the manual.
MD trajectories
MD trajectories are a common data type and are handled with the molecular_trajectory class in MLatom. They consist of molecular_trajectory_step objects which contain information about molecule and other information such as step number, time, energies, etc.:
traj = ml.data.molecular_trajectory()
...
print(f'Info for MD trajectory step {traj.steps[5].step}'')
print(f'Molecule: {traj.steps[5].molecule}')
print(f'Time: {traj.steps[5].time}')
print(f'Kinetic energy: {traj.steps[5].molecule.kinetic_energy}')
The trajectories can be dumped and loaded as different formats: json (easier to open and browse, but heavy and slow, dumping overwrites the file), h5md (faster, smaller, dumping appends, not overwrites), plain text (xyz coordinates, velocities, energies, temperature, etc., dumping overwrites):
traj.dump(filename='mytraj.h5md', format='h5md')
traj2.load(filename='mytraj.h5md', format='h5md')
To unlock many of the features available in molecular databases, it is often convenient to convert the trajectory into molecular_database:
moldb = traj.to_database()
Visualizing data objects
You can use the common tools for visualizing XYZ files, etc. Additionally, MLatom provides several handy routines for quick checks of data objects in Jupyter notebooks (currently, only on the XACS cloud):
my_molecule.view()
my_molecular_database.view()
my_trajectory.view()
and also for vibrations, see below.
Visualizing vibrations
There are several ways to visualize the vibrations:
Currently only on the XACS cloud computing:
At the end of the calculations via input file/command line using the options
freq,ir, orraman, MLatom will dump thefreq_gaussian{mol_index}.logfile, which uses Gaussian-style output format for frequencies. You can open it in ChemCraft.You can also dump text output file using Gaussian-style format for frequencies from the Python API:
mymolecule.dump(filename='gauss.log', format='gaussian').
You can directly view the vibrations in
Jupyter notebookby usingmymolecule.view(normal_mode=...), as shown below for an examplemolvibr.jsonfile:
import mlatom as ml
# let's load the molecule with the required properties
molvibr = ml.molecule()
molvibr.load(filename='molvibr.json')
# choose the normal mode to visualize
normal_mode = 1
# let's view it
molvibr.view(normal_mode=normal_mode)
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
# we can also check the information for this normal mode
print(f'Frequency: {molvibr.frequencies[normal_mode]:.2f} cm^-1')
print(f'Normal mode in Angstrom')
for atom in molvibr.atoms:
disp = atom.normal_modes[normal_mode]
print(f" {disp[0]:8.3f} {disp[1]:8.3f} {disp[2]:8.3f}")
Frequency: 288.70 cm^-1
Normal mode in Angstrom
-0.000 0.000 -0.025
-0.085 0.168 -0.141
0.085 -0.168 -0.141
0.000 0.000 0.184
-0.000 -0.000 -0.003
0.018 -0.021 -0.014
-0.018 0.021 -0.014
0.000 0.000 0.085
-0.000 -0.000 -0.890
# the non-displaced geometry for reference
print(molvibr.get_xyz_string())
9 C -1.2121068000000 -0.2252488500000 0.0000164100000 H -1.2701429600000 -0.8599818400000 -0.8855315500000 H -1.2701264500000 -0.8599834500000 0.8855643400000 H -2.0710636400000 0.4504289000000 0.0000252100000 C 0.0804149500000 0.5556635800000 0.0000041500000 H 0.1395635900000 1.1970970800000 -0.8856250600000 H 0.1395848700000 1.1970854800000 0.8856411200000 O 1.1428329300000 -0.3971737900000 -0.0000106600000 H 1.9796722200000 0.0702558200000 -0.0001121300000
# non-displaced geometry
molvibr.view()
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol