概览
MLatom是一个专注于人工智能增强计算化学的程序包,旨在运用机器学习的强大能力,加快常见模拟的速度并提升其准确性,同时支持构建复杂且高效的工作流程。
用户可以使用输入文件、Python脚本和命令行选项来运行模拟。
MLatom is an open-source package that can be installed via pip and cloned from GitHub. Additional advanced features can be obtained via the Aitomic add-ons. The calculations with MLatom can be performed on the Aitomistic Hub and XACS cloud computing service.
计算化学家能够利用MLatom计算 能量 和 热化学 性质,进行极小值和过渡态的 几何结构优化 ,运行多种类型的 分子动力学模拟 (包括NVE、NVT、 准经典 和 表面跳跃轨迹),传播 量子 动力学,执行扩散蒙特卡洛模拟,以及利用 机器学习、量子力学和组合模型 模拟 (旋转)振动 (包括 红外 和 拉曼光谱)、 单光子紫外/可见光吸收 和 双光子吸收光谱 。用户可以从包含通用机器学习模型 universal ML models ( 我们推荐使用 UAIQM ,但同样支持AIQM1、ANI-1ccx、ANI-1xnr、AIMNet2、DM21等其他模型)的广泛方法库中做出选择;UAIQM、AIQM1和ANI-1ccx能够以低于密度泛函理论(DFT)的成本实现接近耦合簇方法的精度。开发者可以使用各种机器学习算法构建自己的模型,或者使用标准的量子力学方法(密度泛函理论、从头算方法 和半经验方法)。该软件在一定程度上还支持 周期性边界条件。MLatom的极大灵活性在很大程度上得益于其广泛使用了与众多尖端软件包和库的接口。
以下视频简要概述了MLatom的功能:
为帮助您快速入门,我们提供了一个 简单示例 以展示如何使用MLatom。
下面我们将详细介绍MLatom的功能。
本页的文本和图表改编自 J. Chem. Theory Comput. 2024, DOI: 10.1021/acs.jctc.3c01203 (在CC-BY 4.0许可下发布;更多参考文献和理论见原文)。关于MLatom及其开发人员的更多信息请访问 MLatom.com 。
深入了解MLatom的功能
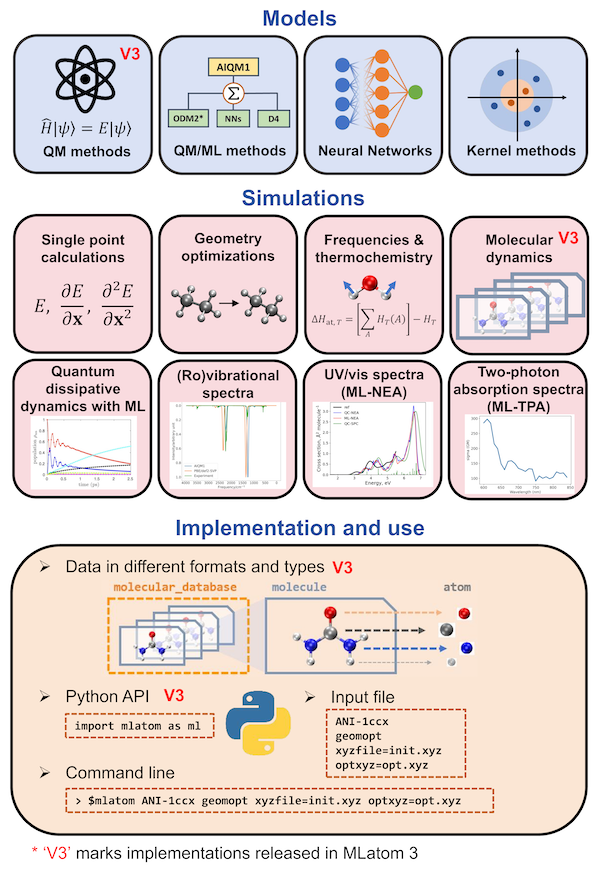
MLatom重点关注分子体系,将典型的量子化学和其他原子模拟软件包的功能与机器学习软件包的功能结合起来。用户可以从一系列现成的QM和ML模型中选择,设计和训练ML模型来执行所需的模拟。MLatom功能的鸟瞰图如下所示:
MLatom作为一个开源包可以通过PyPI安装,即只需使用pip install mlatom或从GitHub上的https://github.com/dralgroup/mlatom获取源代码(参见 安装说明,通常需要额外的包)。为了进一步便捷地使用AI增强的计算化学,MLatom可以在https://XACScloud.com的XACS云计算服务中使用,对于教育和研究等非商业用途,该服务的基本功能是免费的。云计算平台无需安装程序,利于计算资源有限的用户,但是功能的选择可能受到第三方程序可用性的限制(其中一些是商业程序,不能安装在云平台上)。
MLatom支持计算化学家感兴趣的 模拟任务,具有基于 ML、QM及其组合 的通用类型模型。这些任务包括 单点能计算, 几何优化 至最小值和过渡态(可以进行内禀反应坐标(IRC)分析), 频率和热化学性质计算 ,分子动力学 和 量子动力学 , 旋转振动光谱 (红外(IR)和功率)光谱, ML加速的紫外/可见吸收光谱 模拟。MLatom的这一部分更类似于传统的QM和MM软件包,但在模型选择和特殊任务方面具有更大的灵活性。
重要的是,MLatom还允许用户创建、使用和评估自己的ML模型。MLatom支持一系列代表性ML算法,可以作为3D原子结构的函数学习所需的属性。通常,这些算法用于(但不限于)学习势能面,因此为了简单起见,通常可以称为ML(原子间)势(MLPs)。MLatom的一个特殊功能是核脊回归(KRR)算法的原始实现,该算法可用于学习任何属性,作为用户提供的任何输入向量或XYZ分子坐标的函数。此外,用户可以基于Δ-learning、分层ML和自我校正的概念创建自定义的多组件模型。这些模型可能由ML和QM方法组成。MLatom为模型的 训练、 超参数优化 和 评估 提供了标准化的方法,因此从一种模型类型切换到另一种模型类型可能只需要更改一个关键字。这样就可以轻松地试验不同的模型,并为任务选择最合适的模型。
数据 与选择和训练ML算法一样重要。MLatom 提供了几种专门用于计算化学需求的数据结构,主要基于原子、分子、分子数据库和动力学轨迹的通用Python类。这些类不仅允许以清晰的结构化格式存储数据,而且还允许通过转换为不同的分子表示和数据格式,以及将数据集拆分和采样到训练、验证和测试子集中来处理数据。
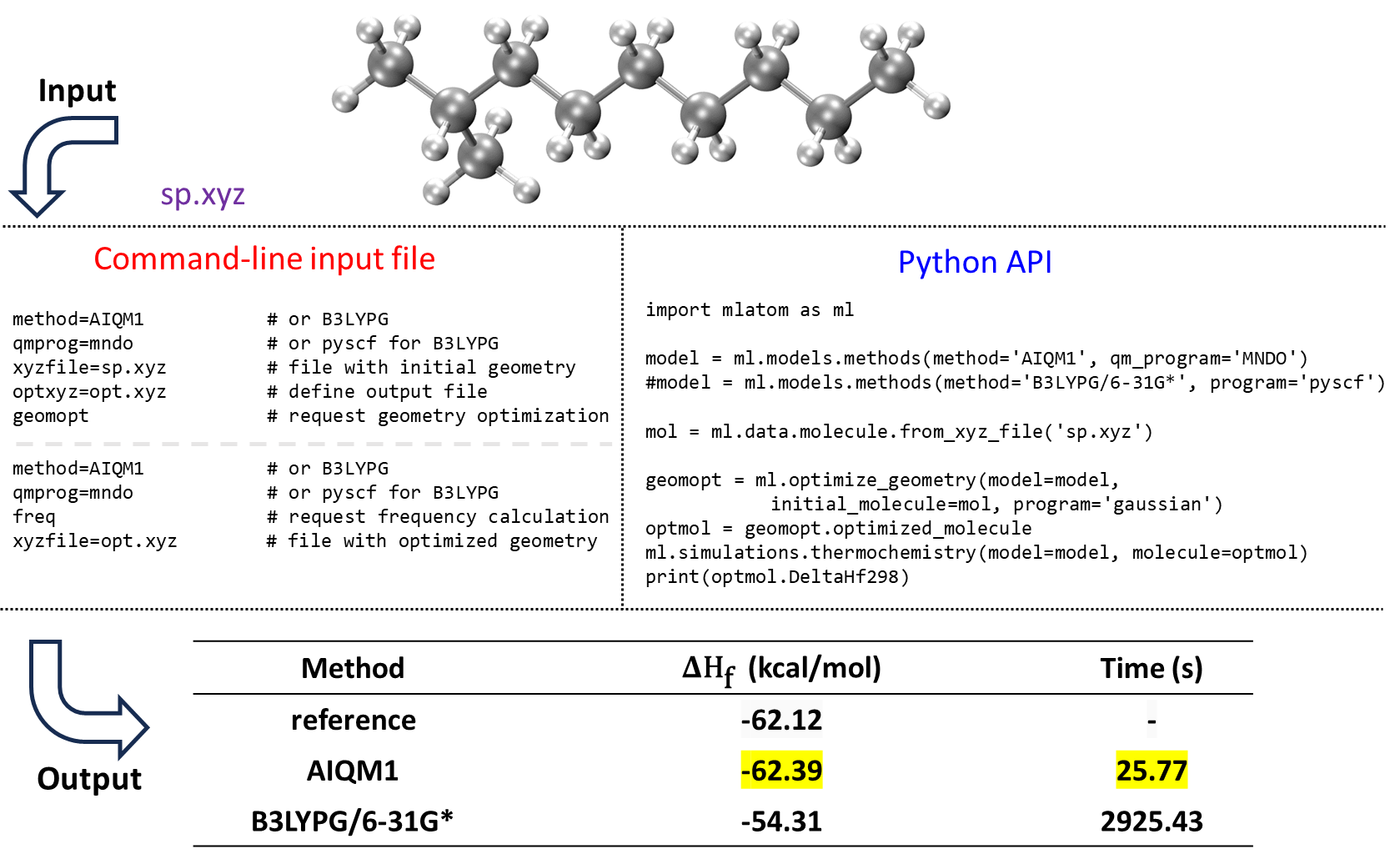
用户与程序的交互方式也很重要,理想情况下,功能应该易于访问并且使用直观。可以直接编辑或提交输入文件、提供命令行选项来进行MLatom计算。另外,MLatom可以作为Python模块导入,并用于创建不同复杂性的计算工作流程。下图给出了这两种方式的并行比较,使用预训练的ML模型ANI-1ccx进行几何优化:
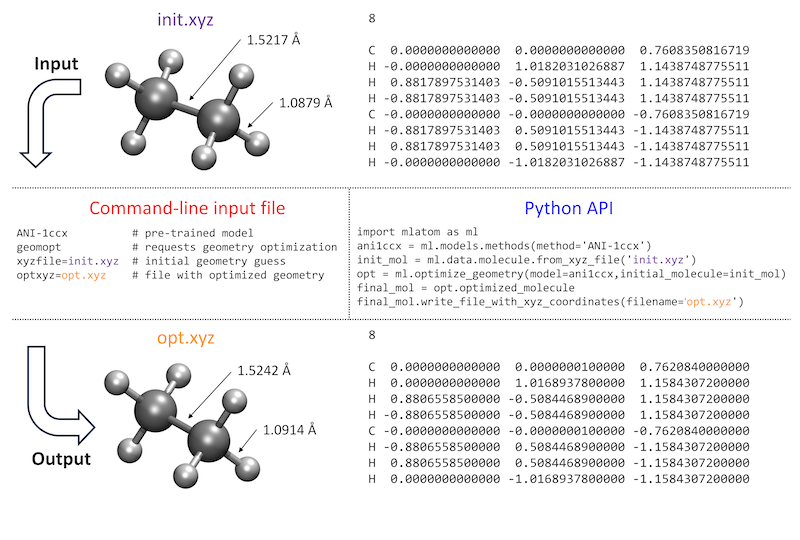
您可以通过下载所需的文件 init.xyz 直接尝试此示例。或者从这里复制粘贴:
8
C 0.0000000000000 0.0000000000000 0.7608350816719
H -0.0000000000000 1.0182031026887 1.1438748775511
H 0.8817897531403 -0.5091015513443 1.1438748775511
H -0.8817897531403 -0.5091015513443 1.1438748775511
C -0.0000000000000 -0.0000000000000 -0.7608350816719
H -0.8817897531403 0.5091015513443 -1.1438748775511
H 0.8817897531403 0.5091015513443 -1.1438748775511
H -0.0000000000000 -1.0182031026887 -1.1438748775511
如果您选择通过命令行选项或输入文件来运行几何优化,你可以从下面下载输入文件 geomopt.inp 或从下面复制粘贴:
ANI-1ccx # pre-trained model
geomopt # requests geometry optimization
xyzfile=init.xyz # initial geometry guess
optxyz=opt.xyz # file with optimized geometry
然后使用以下输入文件运行MLatom模拟:
mlatom geomopt.inp &> geomopt.out
程序会把相关计算信息打印到输出文件 geomopt.out 中。
如果您选择使用PyAPI来运行几何优化,则代码如下:
import mlatom as ml # import MLatom module.
ani = ml.models.methods(method='ANI-1ccx') # denfine an ANI-1cxx model.
init_mol = ml.data.molecule.from_xyz_file('init.xyz') # load the molecular structure.
final_mol = ml.optimize_geometry(model=ani, initial_molecule=init_mol).optimized_molecule # the optimized structure can be obtained by command.
final_mol.xyz_coordinates # we can see the coordinates of the last molecule.
print(final_mol.get_xyz_string()) # print the coordinates in ".xyz" format.
final_mol.write_file_with_xyz_coordinates(filename='opt.xyz') # save it in "opt.xyz".
在本文档中还有更多展示MLatom不同功能的示例。
数据
在MLatom中,一切都围绕于对数据的操作:不同类型的数据库和数据点,如原子、分子、分子数据库和分子轨迹:
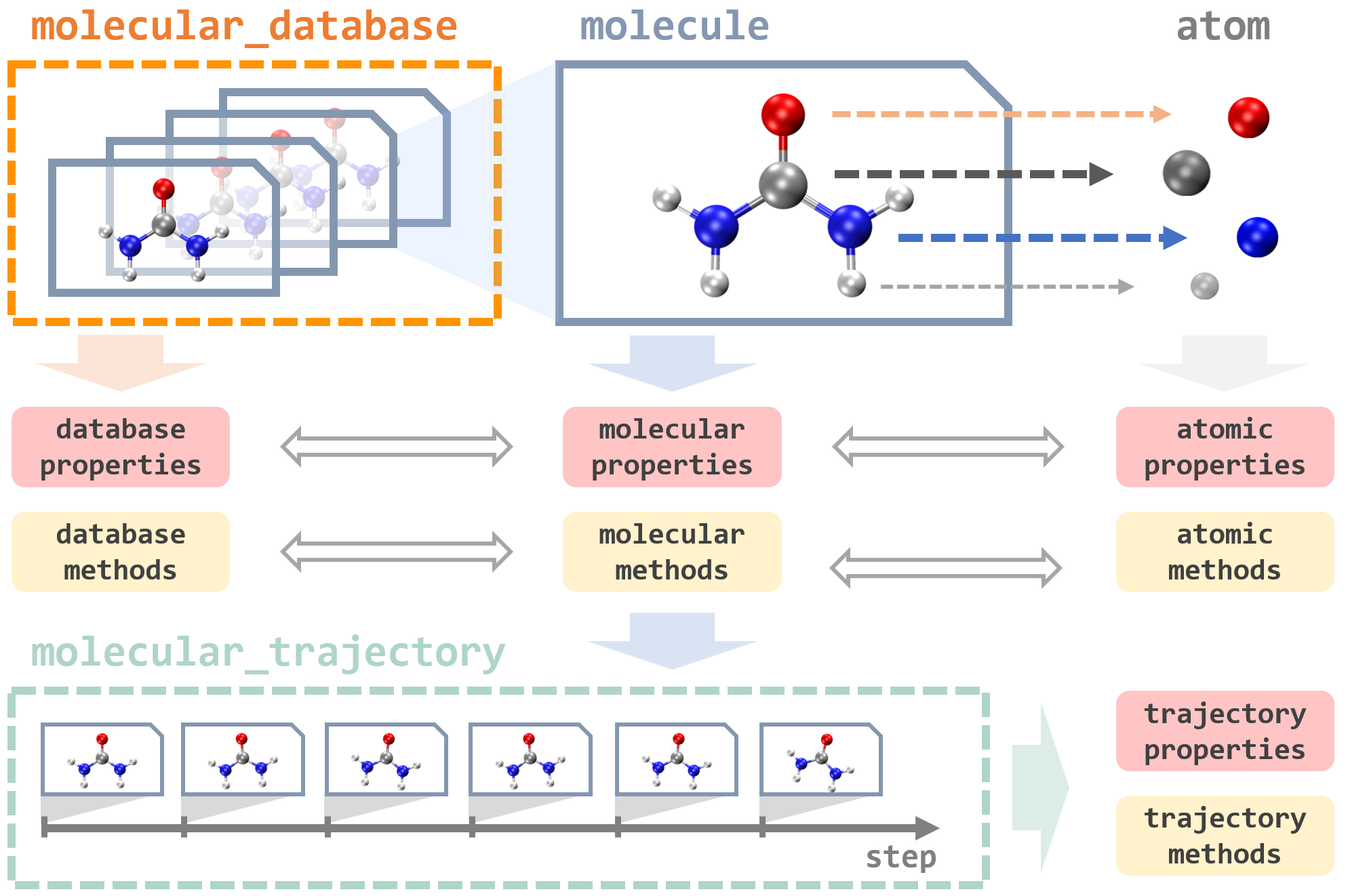
它们被实现为包含许多有用属性的Python类,并提供不同的工具来使用不同的格式加载和转储这些数据类型对象。例如,键类型是一个分子,可以从XYZ文件或SMILES加载,然后自动解析为组成的原子对象。原子对象包含有关核电荷和质量以及核坐标的信息。一个分子对象被赋予电荷和多重度。有关分子和原子性质的信息可以传递给执行模拟,例如分子动力学通过计算量子力学性质(如能量和能量梯度)来更新模型和创建新的分子对象。
在上面的几何优化示例中,从文件 init.xyz 加载了一个分子对象 init_mol 作为几何优化的初始猜测,返回一个优化的构型作为一个新的分子对象 final_mol ,它被保存到 opt.xyz 文件中。数据对象可以通过MLatom Python API直接访问和操作。当在命令行模式下使用MLatom时,许多类似的操作在后台完成,因此用户通常只需要准备标准格式的输入文件,例如带有XYZ坐标的文件。
分子对象可以通过解析分子数据库来组合或创建,该数据库具有将其拆分为训练和验证ML模型所需的不同子集的功能。数据库可以以纯文本(即包括XYZ坐标、标签和XYZ导数的几个文件)、JSON和npz格式加载和转储。另一种数据类型是分子轨迹,它由包含分子和其他信息的时步组成。分子轨迹对象是在几何优化和分子动力学模拟过程中创建的,在后一种情况下,时步是原子动力学轨迹的快照,包含时间、核坐标和速度、原子序数和质量、能量梯度、动能、势能和总能量等信息,如果有的话,还包括偶极矩和其他属性。轨迹可以以JSON、H5MD和纯文本格式加载和转储。提供XYZ坐标的分子可以用几种支持的描述符进行转换:逆核间距离以及相对于平衡结构(RE)、库仑矩阵及其变体的归一化。
MLatom还具有单独的统计例程来计算不同的误差度量并执行其他数据分析。此外,还提供用于准备常见类型图(如散点图和光谱图)的例程。
模型和方法
任何模拟都需要一个为给定输入提供所需输出的模型。模型背后的体系结构和算法可以由用户设计,也可以从已有的选项中选择。ML模型通常需要经过训练才能找到它们的参数,然后才能用于模拟。其中一些模型,如ANI系列的通用MLPs已经进行了预训练。这与QM方法类似,QM方法通常是开箱即用的,不需要调整参数。在MLatom中,我们将任何可以用于模拟的模型称为方法。预训练ML模型和QM方法都属于MLatom术语中的方法,这体现在关键字名称上。该模型类型还包括混合预训练ML和QM方法。MLatom中可用模型的概述:
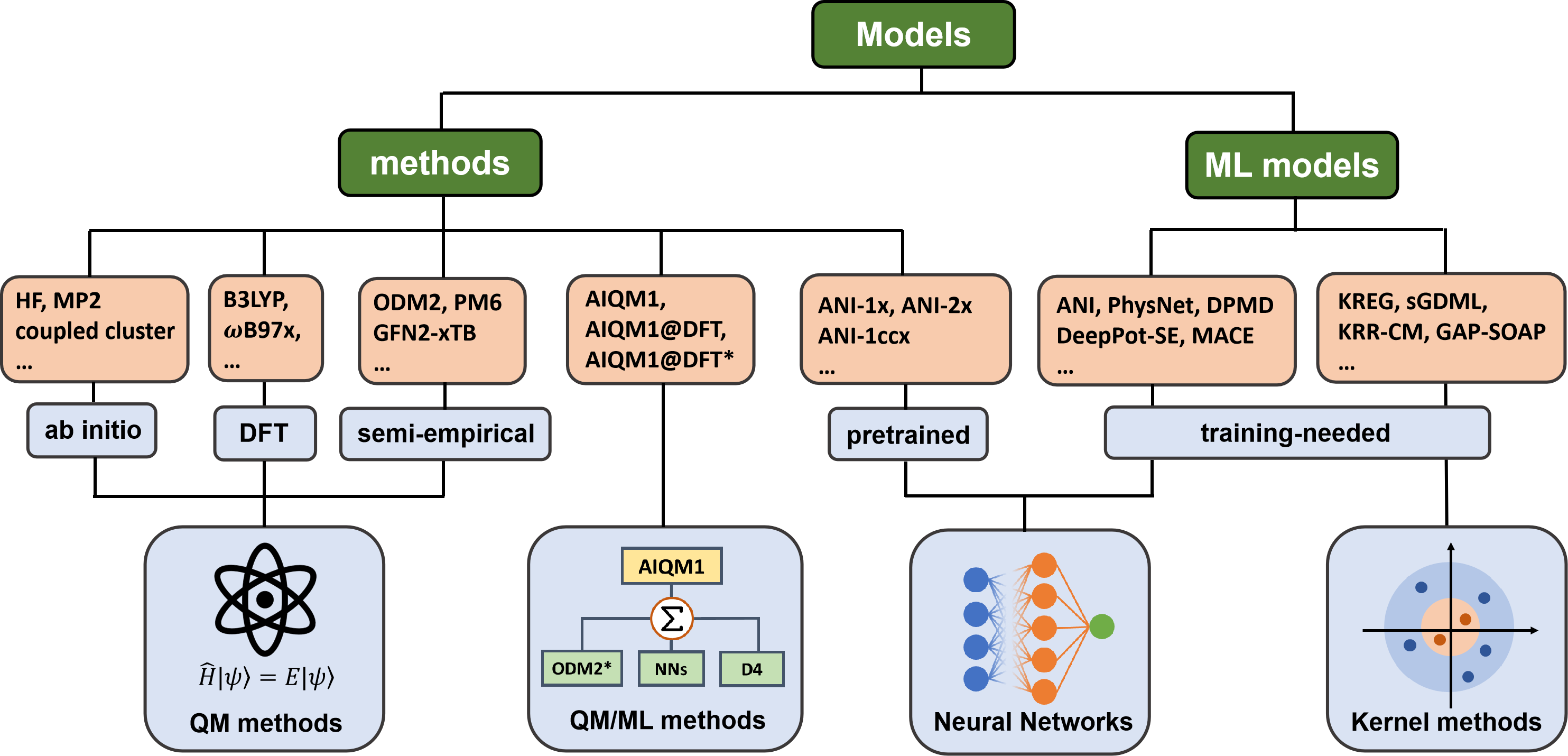
模型实现的概览:
模型类型 |
模型名称 |
实现 |
|---|---|---|
方法 |
(无需训练可直接使用的模型) |
|
QM方法 |
从头算方法,DFT |
|
半经验的OMx, DFTB, NDDO系列方法 |
||
半经验的GFNx-TB方法 |
xtb 的接口 |
|
CCSD(T)*/CBS |
Orca 的接口 |
|
QM/ML方法 |
AIQM1, AIQM1@DFT, AIQM1@DFT* |
|
预训练的ML模型 |
ANI-1x, ANI-2x, ANI-1ccx |
TorchANI 的接口 |
需要训练的模型 |
||
神经网络 |
||
MACE 的接口 |
||
ANI-type |
TorchANI 的接口 |
|
DeePMD-kit 的接口 |
||
PhysNet 的接口 |
||
核方法 |
||
本地实现 |
||
sGDML 的接口 |
||
KRR-CM (带有 库伦矩阵 的KRR) |
本地实现 |
|
GAP套件和 QUIP 的接口 |
方法
MLatom通过与许多第三方的、最先进软件包的接口提供了对多种方法的访问:
预训练的ML模型:
通用势ANI-1ccx、ANI-1x、ANI-2x、ANI-1x-D4和ANI-2x-D4。ANI-1ccx是最准确的,接近CCSD(T)精度。图2展示了其在几何优化中的应用示例。其他方法接近密度泛函理论(DFT)水平。ANI-1ccx和ANI-1x仅限于CHNO元素,而ANI-2x可用于CHNOFCIS元素。用户可以使用D4色散校正的通用ANI电位,这可能对非共价复合物有用。对ωB97X函数进行D4校正,用于生成预训练ANI-1x和ANI-2x的数据。ANI模型通过TorchANI接口提供,D4修正通过dftd4接口提供。这些方法仅限于预测中性闭壳层分子在基态下的能量和力。MLatom会报告使用这些方法计算的不确定度,这些不确定度基于神经网络(NN)预测间的标准差。
用于预测双光子吸收(TPA)截面的特殊ML-TPA模型。
混合QM/ML方法AIQM1, AIQM1@DFT和AIQM1@DFT*比预训练的ML模型更具可转移性和准确性,但速度较慢(介于半经验QM方法和DFT之间,仍然比DFT快得多)。AIQM1接近CCSD(T)精度,而AIQM1@DFT和AIQM1@DFT*针对基态下中性闭壳层分子的DFT精度。所有这些方法都局限于CHNO元素。AIQM1和AIQM1@DFT为ωB97X泛函引入了显式的D4色散修正,而AIQM1@DFT*没有。混合QM/ML方法还包括改进的ANI系列网络和改进的半经验QM方法ODM2(ODM2*,由MNDO或Sparrow提供)。这些方法也可用于计算带电物种、自由基、激发态以及其他量子力学性质(如偶极矩、电荷、振子强度和非绝热耦合)。MLatom根据NN预测之间的标准差报告了使用这些方法进行计算的不确定性。
一系列已建立的QM方法,由从头算方法(例如HF、MP2、耦合簇等)到DFT方法(例如B3LYP、ωB97X等),通过PySCF,Gaussian和Orca接口提供。
一系列半经验QM方法(GFN2-xTB, OM2, ODM2, AM1, PM6等),通过xtb、MNDO和Sparrow接口提供。
一种特殊的复合方法CCSD(T)*/CBS,通过与Orca的接口将CCSD(T)外推到完全集。该方法快速、准确,允许用户使用其他方法检查计算质量,并为ML生成鲁棒的参考数据。该方法用于生成AIQM1和ANI-1ccx的参考数据。
需要训练的可用标准模型
MLPs领域的模型非常丰富。因此用户通常可以选用文献中报道的流行MLP体系结构之一,而无需开发新的MLP。MLatom提供了不同类型MLP的工具集。这些支持的类型大致分为以下两类:
基于神经网络(NNs)的、具有固定的局部描述符的模型(ANI系列MLPs和DPMD属于固定的局部描述符),和以PhysNet和DeepPot-SE为代表的学习的局部描述符。MLatom还支持具有代表性的等变NN MACE,它在许多任务中展现出优越的性能。
基于核方法(KMs)的、具有全局描述符的模型(KREG、sGDML和KRR-CM属于全局描述符),以及仅由GAP-SOAP表示的局部描述符。
这些模型中的任何一个都可以 训练 并用于模拟,例如几何优化或动力学。MLatom还支持许多算法的超参数优化,包括网格搜索、通过hyperopt包进行的Bayesian优化,以及SciPy中支持的标准优化算法。优化后的模型的泛化误差也可以用标准的方法进行评估(保留和交叉验证)。
基于核方法的自定义模型
MLatom还支持训练基于核脊回归(KRR)的自定义模型,用于给定一组输入向量x或XYZ坐标和任何标签y。如果提供了XYZ坐标,它们可以在几个支持的描述符之一中进行转换(例如逆核间距离以及相对于平衡结构(RE)、库仑矩阵及其变体的归一化)。用户可以从实现的核函数中选择一种,包括线性、高斯、指数、拉普拉斯和Matérn,以及周期和衰减周期函数,如表2所示。这些核函数 \(k(\mathbf{x},\mathbf{x}_j;\mathbf{h})\) 是解决求近似函数 \(f(\mathbf{x};\mathbf{h})\) 的回归系数 \(\alpha\) 的KRR问题所需的关键分量的输入向量 \(\mathbf{x}\) :
可用于解决核脊回归问题的核函数总结:

在大多数情况下,核函数有超参数h需要调整,它们可以被视为测量输入向量 \(\mathbf{x}\) 和所有 \(N_\text{tr}\) 训练点 \(\mathbf{x}_j\) 之间的相似性(两个向量的长度 \(N_x\) 应该是相同的)。除了核函数中的超参数外,所有KRR模型至少还有一个正则化参数 \(\lambda\) ,即在训练期间使用的用于提高泛化性的参数。
复合模型
通常,将几个模型结合起来是有益的。这种复合模型的一个例子是基于Δ-learning,低级QM方法被用作基线,通过ML模型对其进行校正,以接近目标高级QM方法的准确性。另一个例子是集成学习,创建了个ML模型,在模拟期间对它们的预测进行平均,以获得更鲁棒的结果,并用于 主动学习 的query-by-committee策略。这两个概念也可以在更复杂的工作流程中结合,例如 AIQM1 方法使用神经网络集合作为校正Δ-learning模型,半经验QM方法作为基线。为了便捷地实现这些工作流程,MLatom允许将组合模型构建为模型树 model_tree_node,参见AIQM1的示例:
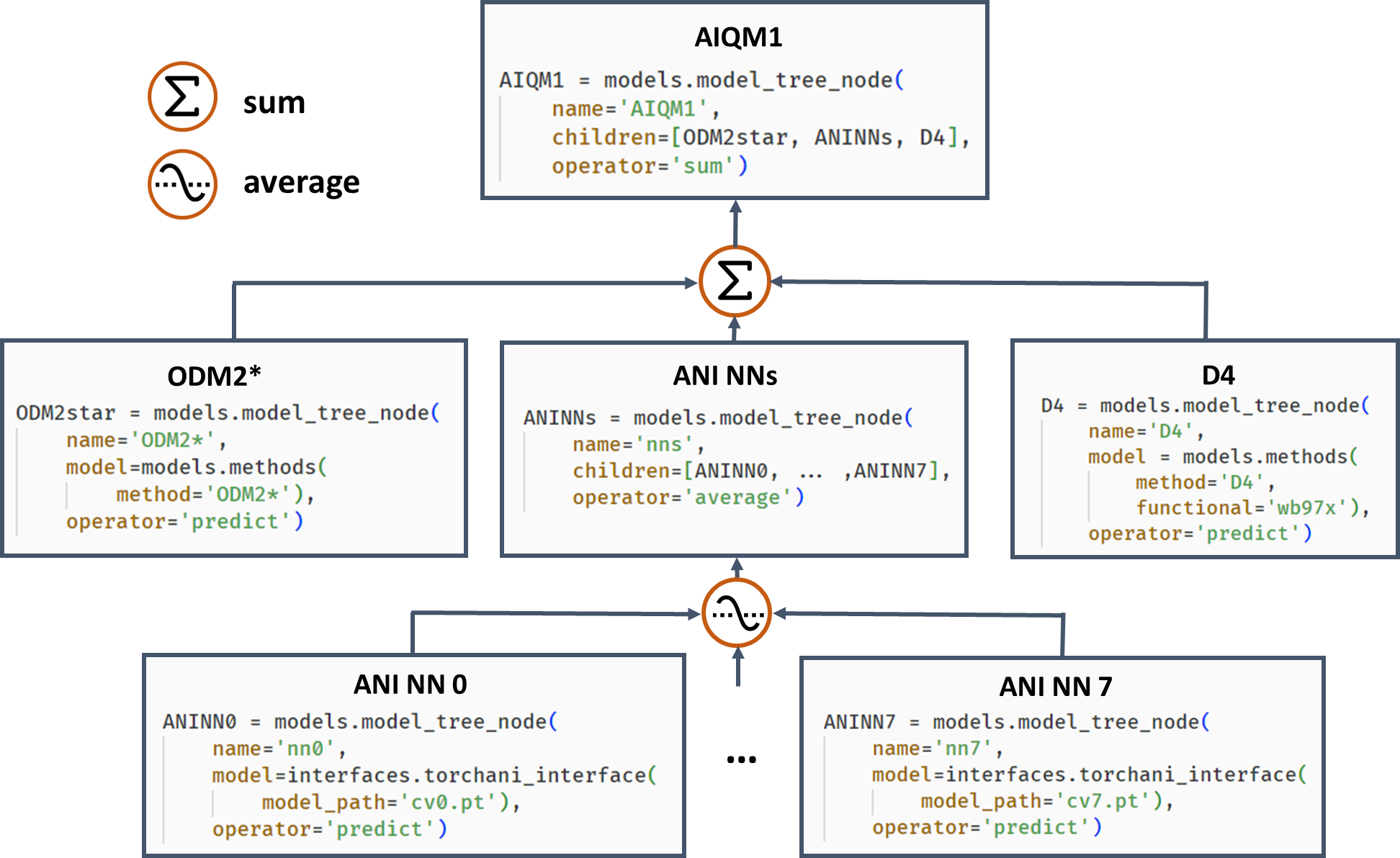
AIQM1的根父节点包括3个子节点,即半经验QM方法ODM2*、NN集合和额外的D4色散校正。NN集合又是8个ANI类型NN子节点的父节点。通过将操作“average”或“sum”应用于子节点的预测,可以获得父节点的预测。代码片段也显示了出来。
其他组合模型的例子是分层ML,每当下一个ML模型校正前一个模型的预测时,会结合几个(校正)ML模型(这些模型的训练基于QM水平间的差异)以及自我校正。
模拟
MLatom支持一系列模拟任务,如 单点能计算 、 几何优化 、 频率和热化学计算 、 分子动力学 和 量子动力学 、 单光子 和 双光子吸收 以及 (旋转)振动光谱 模拟。它们大多需要任何能够提供能量和能量导数(梯度和Hessian函数)的 模型 。
单点能计算
单点能计算是对量子力学性质的计算——主要是对能量和能量梯度的计算,也包括对单一几何构型的Hessian、电荷、偶极矩等的计算。这些计算在计算化学的ML研究中非常常见,因为它们可用于使用QM方法生成用于训练和验证ML的参考数据、使用ML进行推断以验证训练模型并生成新几何构型所需的数据。MLatom是一个便捷的工具,不仅可以像许多QM包一样对单个几何构型执行单点能计算,还可以对具有许多几何构型的数据集执行单点能计算(请参阅专用 教程)。
几何优化
定位势能面(PES)上的驻点(如能量最小值和过渡态)对于理解分子结构和反应性至关重要。因此,几何优化是计算化学中最重要和最常见的任务之一。MLatom可以使用任何提供能量和梯度的模型定位能量最小值和过渡态(TS)。几何优化的一个例子是在“开始”给出的,也可参阅专用 教程 。MLatom的一个实际应用是,此前对环对苯乙烯(CPP)纳米材料及其配合物与富勒烯分子(体系有多达200个原子)这样的大体系进行了高效和精确的几何优化:
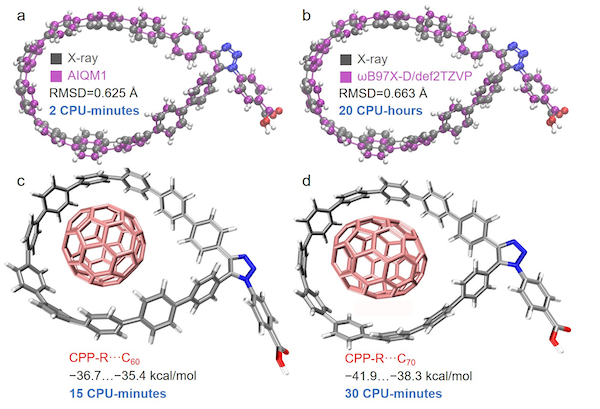
功能化环丙苯(CPP)纳米套索的X射线结构与在真空中优化的结构重叠,分别使用(a) AIQM1和(b) ωB97X-D/def2-TZVP进行计算(这两种计算均可通过MLatom进行,详见 教程)。使用AIQM1方法计算得到功能化的CPP与(c) C60 和(d) C70 的复合物在真空中的结合能(以kcal/mol为单位),同时还提供了这些计算所需的CPU时间。
AIQM1方法能够提供经过优化的功能化CPP结构,其与X射线结构的符合度比从DFT方法得到的结构更好,并且速度比DFT方法快600倍。在我们的实验室中,我们还使用AIQM1方法在单个CPU上优化拥有一千多个原子的体系,而对于计算更密集的任务,如大体系的动力学,可以使用预训练的ANI方法。Berny TS优化算法还需要Hessian。一旦定位了TS,用户可以按照内禀反应坐标(IRC)来检查其性质。
几何优化可以使用SciPy、ASE或Gaussian接口提供的多种算法进行。在ASE中可以使用dimer方法进行TS搜索,在Gaussian中可以使用Berny算法进行TS搜索。IRC计算仅支持使用Gaussian接口。
将各种QM和ML方法进行几何优化的无缝集成具有优势,因为它允许使用其本身无法实现的某些模拟任务的接口程序中的方法。例如,MLatom可以通过与xtb程序的接口使用GFN2-xTB方法进行TS搜索,而后者程序本身没有TS搜索选项。类似地,Sparrow提供对许多半经验方法的访问,但只能用于单点能计算。由于许多模型和实现中都没有提供解析梯度和Hessian,MLatom还实现了有限差分数值微分,进一步扩展了这些模型在几何优化中的适用性。
频率计算
模拟振动频率是计算化学中另一个常见且重要的任务,因为它有助于进一步验证驻点的性质,可视化分子振动,计算零点振动能(ZPE)和热化学性质,并获取能够与实验振动光谱进行比较的光谱信息。这些计算可以通过经调整的TorchANI实现、Gaussian接口和PySCF接口在脊转子谐波近似内进行。Gaussian界面也允许使用二阶微扰方法计算非谐波频率。
与几何优化类似,MLatom可以使用任何提供能量的模型——包括ML和QM或其组合进行 这些模拟 。计算还需要Hessian,并且在可能的情况下,会使用解析Hessian。如果解析Hessian不可用,可以计算半解析(带有解析梯度)或完全数值的Hessian。
相对能量计算
相对能量对于理解和预测化学行为的各个方面至关重要,从动力学到热力学,例如通过计算反应能、势垒高度、异构化能以及分子稳定性。MLatom可以为分子生成各种类型的能量,如零点振动能、总能量、焓、熵、吉布斯自由能和内能。因此,该软件包可以用于评估不同类型的相对能量,例如上述用于研究哪些富勒烯分子与环丙苯纳米套索结合更强、环戊二烯和马来酰亚胺的Diels–Alder反应的反应焓和吉布斯自由能。
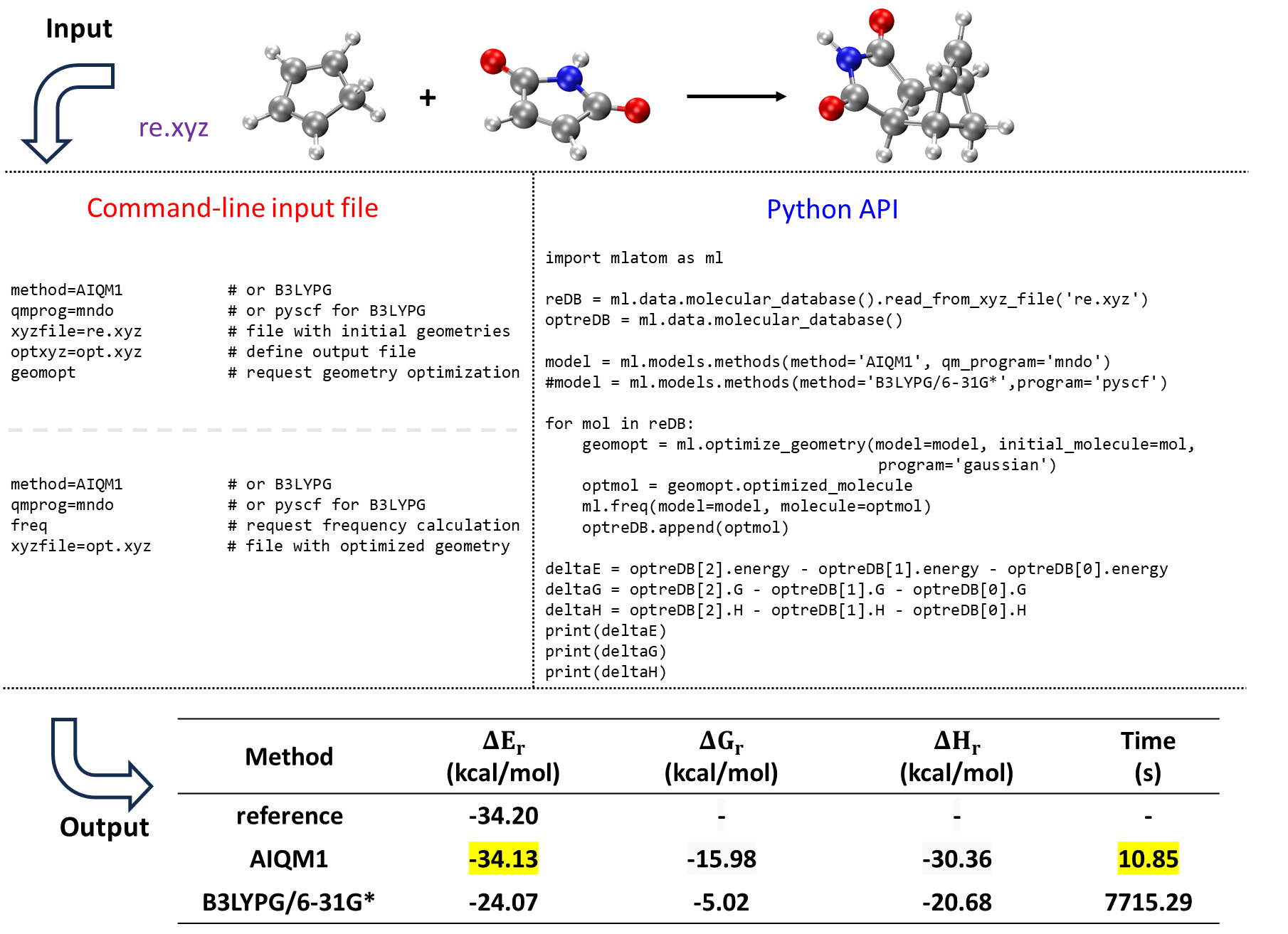
在这个例子中,使用AIQM1和B3LYPG/6-31G*(通过与PySCF的接口;B3LYPG中的“G”表示我们按照Gaussian程序的约定使用B3LYP变体)计算环戊二烯和马来酰亚胺进行Diels–Alder反应形成相应内端产物的零点振动能排斥能、吉布斯自由能和焓变化。参考反应能来自GMTKN55数据集。
我们将展示使用Python API进行此操作的便捷性(通过手动输入文件/命令行进行计算,为每个分子执行热力学计算,并使用上图所示选项获取每种能量类型的变化,也可以完成相同的操作)。
import mlatom as ml
# read molecules from .xyz file
reDB = ml.data.molecular_database().read_from_xyz_file('re.xyz')
optreDB = ml.data.molecular_database()
# define method to calculate thermochemical properties
model = ml.models.methods(method='AIQM1', qm_program='mndo')
for mol in reDB:
# geometry optimization
geomopt = ml.optimize_geometry(model=model, initial_molecule=mol, program='gaussian')
optmol = geomopt.optimized_molecule
# frequency calculation
ml.thermochemistry(model=model, molecule=optmol)
optreDB.append(optmol)
# get relative energies
deltaE = optreDB[2].energy - optreDB[1].energy - optreDB[0].energy
deltaG = optreDB[2].G - optreDB[1].G - optreDB[0].G
deltaH = optreDB[2].H - optreDB[1].H - optreDB[0].H
print(deltaE)
print(deltaG)
print(deltaH)
输出如上图所示。
备注
为发挥AIQM1的全部潜力,我们建议安装MNDO程序作为其QM部分的支持。由于许可原因,XACS云使用Sparrow,但目前它仅提供数值梯度,限制了其在几何优化中的适用性。因此,您可以尝试在XACS云上直接使用ANI-1ccx,通常它是一个非常好的替代选择(尽管它仅限于基态中的中性闭壳层化合物)。
同样,在XACS云平台上,您可以尝试使用ASE作为优化器,而非Gaussian。
生成热的计算
相对能量计算的一种特殊类型是生成热(焓)的评估。MLatom使用类似于从头算和半经验QM计算中使用的方案来推导生成热:
其中 \(\Delta H_{\text{f},T}(A)\) 为自由原子 \(A\) 的实验生成焓, \(\Delta H_{\text{at},T}\) 为温度 \(T\) 下的原子化焓。
该方案需要知道自由原子能量E(A)。任何能够计算这些能量的模型都可以用于预测生成热。对于QM方法,这显然可以直接进行,对于ML模型,如果孤立原子的能量包含在训练数据中,也可进行预测。然而,如果基于ML的模型仅在分子物种上进行训练(这是常见的做法),不能期望它们产生合理的生成热。在MLatom支持的预训练模型的情况下,我们以前为AIQM1和ANI-1ccx方法拟合了自由原子能量,以重现一组常见分子的实验生成热,因为这些方法中的神经网络并未在孤立原子上进行训练。结果表明,这两种方法都可以提供 接近化学精度 的生成热,速度比替代的高精度QM方法快几个数量级。此外,我们提供了一种基于这些方法中NN预测偏差的不确定性量化方案,以告知用户预测的可信度,这对于发现生成热的实验数据集中的错误非常有用。
使用MLatom计算AIQM1和B3LYP/6-31G*方法计算2-甲基辛烷生成热的示例:
通过将计算值与实验进行比较,可以看出AIQM1更快更准确,相比之下,B3LYP的性能较差。这也与我们先前的基准测试结果一致。
以下是计算较小的乙醇分子在ANI-1ccx水平的生成热的输入文件示例:
freq # 1. requests frequency calculation
ANI-1ccx # 2. pre-trained model, the same as that used in geometry optimization
xyzfile=opt.xyz # 3. file with optimized geometry
用户可以直接使用我们提供的预先优化好的几何构型( opt.xyz ):
9
C -1.691449880 -0.315985130 0.000000000
H -1.334777040 0.188413060 0.873651500
H -1.334777040 0.188413060 -0.873651500
H -2.761449880 -0.315971940 0.000000000
C -1.178134160 -1.767917280 0.000000000
H -1.534806620 -2.272315330 0.873651740
H -1.534807450 -2.272316160 -0.873650920
O 0.251865840 -1.767934180 -0.000001150
H 0.572301420 -2.672876720 0.000175020
当用户创建好输入文件以后可以运行MLatom,例如在XACS云平台上进行:
mlatom freq.inp &> freq.out
程序的输出会保存在 freq.out 文件中。
或者,您可以在命令行中使用相同的选项运行相同的模拟:
mlatom freq ANI-1ccx xyzfile=opt.xyz
在MLatom的输出中,用户可以找到振动分析和热化学结果。振动分析会输出每个简振模式的频率、约化质量和力常数。热化学部分会打印出零点能、焓、吉布斯自由能、生成热和其他性质。在本示例中,输出如下所示:
==============================================================================
Vibration analysis for molecule 1
==============================================================================
Multiplicity: 1
Rotational symmetry number: 1
This is a nonlinear molecule
Mode Frequencies Reduced masses Force Constants
(cm^-1) (AMU) (mDyne/A)
1 261.0407 1.0929 0.0439
2 286.9474 1.1295 0.0548
3 428.0328 2.7260 0.2943
4 841.1644 1.0947 0.4564
5 928.2688 2.3346 1.1853
6 1082.6338 3.1779 2.1946
7 1138.6399 1.9583 1.4959
8 1212.5257 1.5518 1.3442
9 1287.5002 1.1134 1.0874
10 1292.8263 1.0682 1.0520
11 1426.7497 1.2307 1.4760
12 1457.4408 1.3203 1.6524
13 1496.9454 1.1496 1.5178
14 1501.2654 1.0458 1.3887
15 1524.8936 1.0529 1.4426
16 3063.7060 1.0530 5.8232
17 3076.9020 1.1120 6.2026
18 3093.2039 1.0342 5.8299
19 3201.4189 1.1000 6.6424
20 3213.8757 1.1004 6.6964
21 3741.4822 1.0681 8.8098
==============================================================================
Thermochemistry for molecule 1
==============================================================================
Standard deviation of NNs : 0.00063190 Hartree 0.39652 kcal/mol
Energy : -154.89195912 Hartree
ZPE-exclusive internal energy at 0 K: -154.89196 Hartree
Zero-point vibrational energy : 0.08101 Hartree
Internal energy at 0 K: -154.81095 Hartree
Enthalpy at 298 K: -154.80576 Hartree
Gibbs free energy at 298 K: -154.83631 Hartree
Atomization enthalpy at 0 K: 1.21200 Hartree 760.54458 kcal/mol
ZPE-exclusive atomization energy at 0 K: 1.29301 Hartree 811.37653 kcal/mol
Heat of formation at 298 K: -0.09030 Hartree -56.66187 kcal/mol
分子动力学
MLatom拥有原生的分子动力学(MD)实现,支持任何提供力的模型,不一定需要是保守的(请参阅 教程)。通常,基于QM方法的计算可以采用称为从头算或BOMD的变体。ML势能的普及使得可以以与分子力场相当的成本或比常用的基于DFT的BOMD快得多的速度执行BOMD质量的动力学模拟,这使用户可以对大体系进行常规模拟,例如含有大约400个原子的八烷基四烯桥接的邻苝二酰亚胺的四联组装:
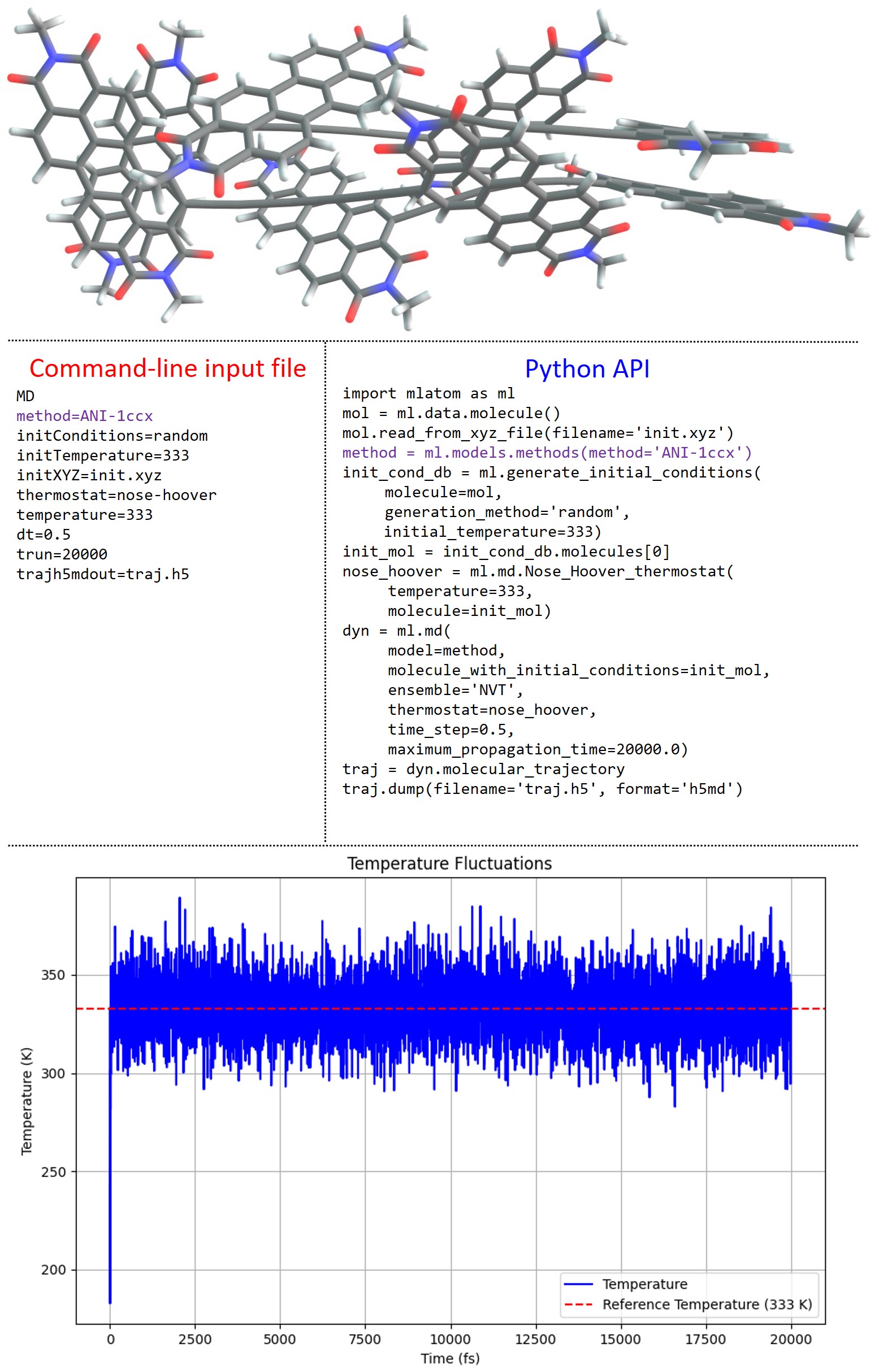
这类模拟的准确性也可以很高;例如使用AIQM1方法进行的MD得到的红外光谱比速度慢得多的DFT MD得到的光谱更准确(见下文)。
目前支持基于速度Verlet算法的NVE和NVT系宗的模拟。可以使用Andersen和Nosé–Hoover恒温器进行NVT模拟,预计未来将提供其他恒温器的实现。轨迹可以以不同的格式保存,包括纯文本、JSON和更紧凑的H5MD数据库格式。此外,我们将Fortran中实现的KREG模型更好地集成到主要基于Python的MLatom代码中,使得KREG模型的MD非常高效。
输入文件 h2_md_kreg.inp 展示了如何使用Nosé–Hoover恒温器在NVT中对氢分子进行动力学模拟:
# h2_md_kreg.inp
MD # 1. requests molecular dynamics
initConditions=user-defined # 2. use user-defined initial conditions
initXYZ=h2_md_kreg_init.xyz # 3. file with initial geometry; Unit: Angstrom
initVXYZ=h2_md_kreg_init.vxyz # 4. file with initial velocity; Unit: Angstrom/fs
dt=0.3 # 5. time step; Unit: fs
trun=30 # 6. total time; Unit: fs
thermostat=Nose-Hoover # 7. use Nose-Hoover thermostat
ensemble=NVT # 8. NVT ensemble
temperature=300 # 9. Run MD at 300 Kelvin
MLmodelType=KREG # 10. KREG model is used
MLprog=MLatomF # 11. use KREG implemented in the Fortran part of MLatom
MLmodelIn=h2_kreg_energies.unf # 12. file with the trained model
对于本教程,您可以下载具有初始XYZ坐标的 h2_md_kreg_init.xyz 文件,初始XYZ速度的 h2_md_kreg_init.vxyz 文件和模型文件 h2_kreg_energies.unf 。
初始坐标以XYZ格式(Å)定义:
2
H 0.0 0.0 1.0
H 0.0 0.0 0.0
初始速度也以简化的XYZ格式定义(Å/fs):
2
0.0 0.0 -0.05
0.0 0.0 0.05
运行分子动力学模拟。
mlatom h2_md_kreg.inp &> h2_md_kreg.out
模拟结束后,分子的XYZ坐标轨迹将被保存在 traj.xyz 文件中,XYZ速度将被保存在 traj.vxyz 文件中动能、势能和总能(单位Hartree)将被分别保存在 traj.ekin, traj.epot 和 traj.etot 中,能量梯度(单位Hartree/Å)将被保存在 traj.grad 中。
请注意,也可以使用4D时空AI原子模型的概念在没有力的情况下传播MD,该模型直接预测时间函数的核配置。我们对这一概念的实现被称为GICnet模型,目前可在MLatom的一个 公开可用的开发版本 中使用。
上述实现可以在绝热势能面上传播MD,通常用于基态动力学。基于轨迹表面跃迁算法的非绝热MD也可以通过MLatom进行,目前是通过 Newton-X 与MLatom的接口实现的。MLatom还支持量子耗散动力学,如下一节所述。
量子耗散动力学
有时需要将整个体系以量子力学的方式进行处理,并考虑环境效应。这可以通过许多量子耗散动力学(QD)算法完成,并且目前有越来越多的ML技术可用于加速这类模拟。MLatom允许使用基于KRR的递归方案或概念上不同的AI-QD方法执行多个独特的ML加速的QD模拟(请参阅 教程 ),该方法预测轨迹并将其作为时间的函数,或者使用OSTL技术一次性输出整个轨迹。这些方法可以通过MLQD的接口实现。
在递归的KRR方案中,训练了一个KRR模型,建立了未来和过去动力学之间的映射。当向该KRR模型提供当前动力学的简要快照时,可以利用它来预测未来动力学。在AI-QD方法中,训练了一个卷积神经网络(CNN)模型,将仿真参数和时间映射到相应体系状态。使用训练好的CNN模型,可以在不需要显式模拟动力学的情况下预测体系的状态。类似地,超快速的OSTL方法利用基于CNN的架构,根据仿真参数,以单次预测的方式预测体系状态的未来动力学,直到预定义的时间。此外,由于优化是训练的关键组成部分,用户可以使用MLatom的KRR网格搜索功能以及通过hyperopt库的CNN进行Bayesian优化来优化KRR和CNN模型。此外,我们还提供了自动绘图功能,对预测的动力学与提供的参考轨迹进行对比。
振动(红外和功率)光谱
使用MLatom可以通过多种方式计算转动振动光谱。最简单的方法是在优化的分子几何构型上进行频率计算(参见上文和 教程 )。这需要提供Hessian(最好也提供偶极矩)的模型。另一种方法是使用提供能量梯度的模型进行分子动力学模拟,然后处理轨迹(请参阅 手册 )。
无论是频率计算还是基于MD的方法,模型都需要提供偶极矩以计算吸收强度。如果没有提供偶极矩,只能获得频率,或者在MD的情况下,只能获得功率谱而不是红外谱。使用我们的实现,通过对偶极矩的自相关函数进行快速傅里叶变换可以获得红外光谱。功率谱则只需要进行快速傅里叶变换,这在MLatom中也有实现。
我们先前已经展示了AIQM1方法的优点,相比使用DFT(速度慢)或半经验QM方法获得的光谱,从MD模拟中获得的红外光谱相当准确,以下是 N2O 分子的一个示例:

MLatom为每种方法生成光谱;此处对结果进行了整理,并与实验光谱进行比较。
单光子紫外/可见吸收光谱
紫外/可见吸收光谱的模拟需要密集的计算,因为需要计算激发态性质。此外,通过核系宗方法(NEA)可以获得更高质量的光谱,该方法需要为数千个几何构型计算激发态性质以获得高精度。MLatom实现了一个插值ML-NEA方案,提高了光谱的精度,而计算成本仅为传统NEA模拟的一小部分(请参阅 教程):
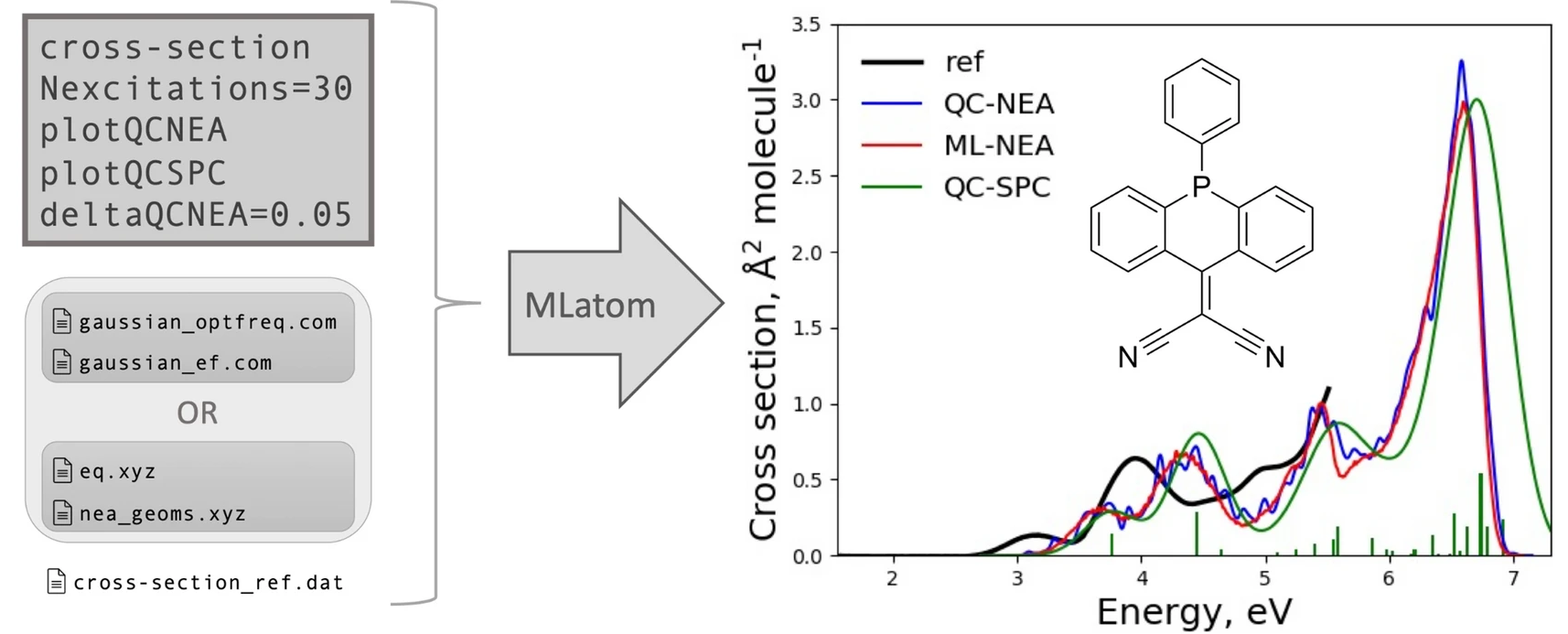
右图所示的ML-NEA预测的截面与传统的QC-NEA和单点卷积方法(QC-SPC)进行了比较。
目前,ML-NEA的计算是基于Newton-X和Gaussian的接口,并利用谐波Wigner分布的几何采样。该方案还能自动确定所需参考计算的最佳数量,提供用户友好的黑盒算法实现。
双光子吸收
除了单光子吸收外,MLatom还实现了一种独特的ML方法,用于基于分子的SMILES字符串计算双光子吸收(TPA)截面,这些字符串通过RDKit接口转换为所需的描述符,并提供溶剂信息。这个ML-TPA方法非常快速,其准确性可与计算成本更高的QM方法相媲美。我们提供了一个在实验数据上预训练的ML模型。ML-TPA在实际的实验室环境中进行了测试,并且对于训练实验数据库中不存在的新分子,也能进行很好的估计。使用ML-TPA预测两光子吸收的示例(请参阅 手册 ):
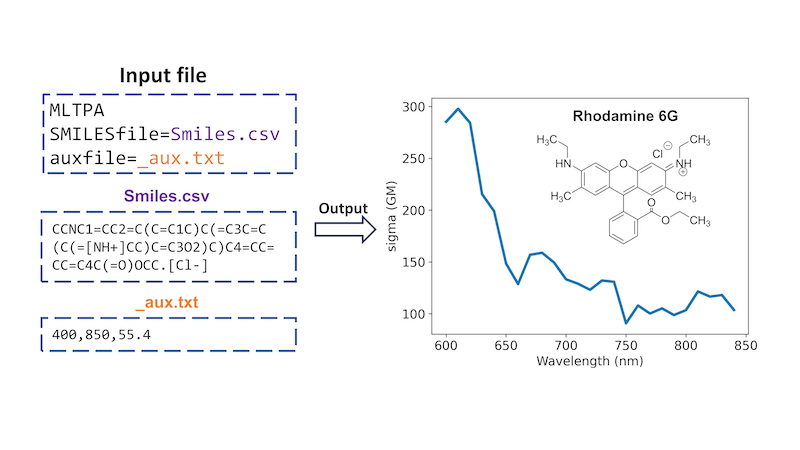
机器学习
我们讨论了支持的 模型 类型以及它们如何应用于 模拟。在这里,我们简要概述了使用MLatom对ML模型进行 训练 和 验证 的一般考虑因素。这些模型共享MLatom的标准约定,包括输入、输出、训练、 超参数优化 和 测试,这可以便捷地在不同模型之间切换并进行基准测试。
训练
要创建一个ML模型,用户必须选择和训练ML模型,并准备数据。MLatom为此过程的不同阶段提供了许多工具。模型可以从提供的各种ML模型类型中选择,这些模型具有预定义的架构,或者可以基于可用的算法和预设模型进行定制。选择了一个模型后,必须对其进行训练,并且在许多情况下,优化其超参数是有益的,甚至是必需的(特别是对于核方法的情况)。
在训练时,应准备适当的数据集。MLatom对数据集拆分有严格的命名规范,以避免在更改和比较不同模型类型时产生混淆。 所有直接或间接用于创建ML模型的数据都称为训练集。 这意味着验证集可用于在NN训练期间进行超参数优化或提前停止,并且是训练集的子集。因此,在排除验证集后剩下的训练集的部分称为 子训练集 ,用于训练模型,即优化模型参数(在NN术语中是权重,在核方法术语中是回归系数)。
MLatom可以将训练数据集拆分为子训练集和验证集,或者通过交叉创建这些子集的集合进行验证。对子集的采样可以随机执行,也可以使用最远点或基于结构的采样。
在核方法下,MLatom中的最终模型通常在超参数优化之后在整个训练集上进行训练。这是可行的,因为核方法对于找到它们的回归系数有一个封闭的解析解,而在选择了适当的超参数之后,过拟合可以在很大程度上得到缓解。在神经网络下,最终模型是在子训练集上训练的,因为在没有任何验证集用于检查过拟合的情况下,在整个训练集上进行训练可能过于危险。
训练预定义的ML模型类型
大多数预定义的ML模型类型,例如ANI系列或KREG模型,都期望以XYZ分子坐标作为输入。这应该由用户提供,或者可以使用MLatom的转换例程获取,例如从SMILES字符串中获取,这依赖于OpenBabel的Pybel API。这些模型具有默认的一组超参数,但特别是对于像KREG这样的核方法,强烈建议进行优化。这些模型原则上可以在任何分子性质上进行训练。它们通常被用于学习势能面,因此在训练集中需要能量标签。如果提供能量梯度进行训练,可以极大地提高PES模型的准确性。因此,通常可以证明增加训练时间是合理的。
对比命令行与Python API中,对尿素分子势能面数据集(从WS22数据库中随机抽样,详见 教程 )上的KREG模型进行训练和测试的用法。
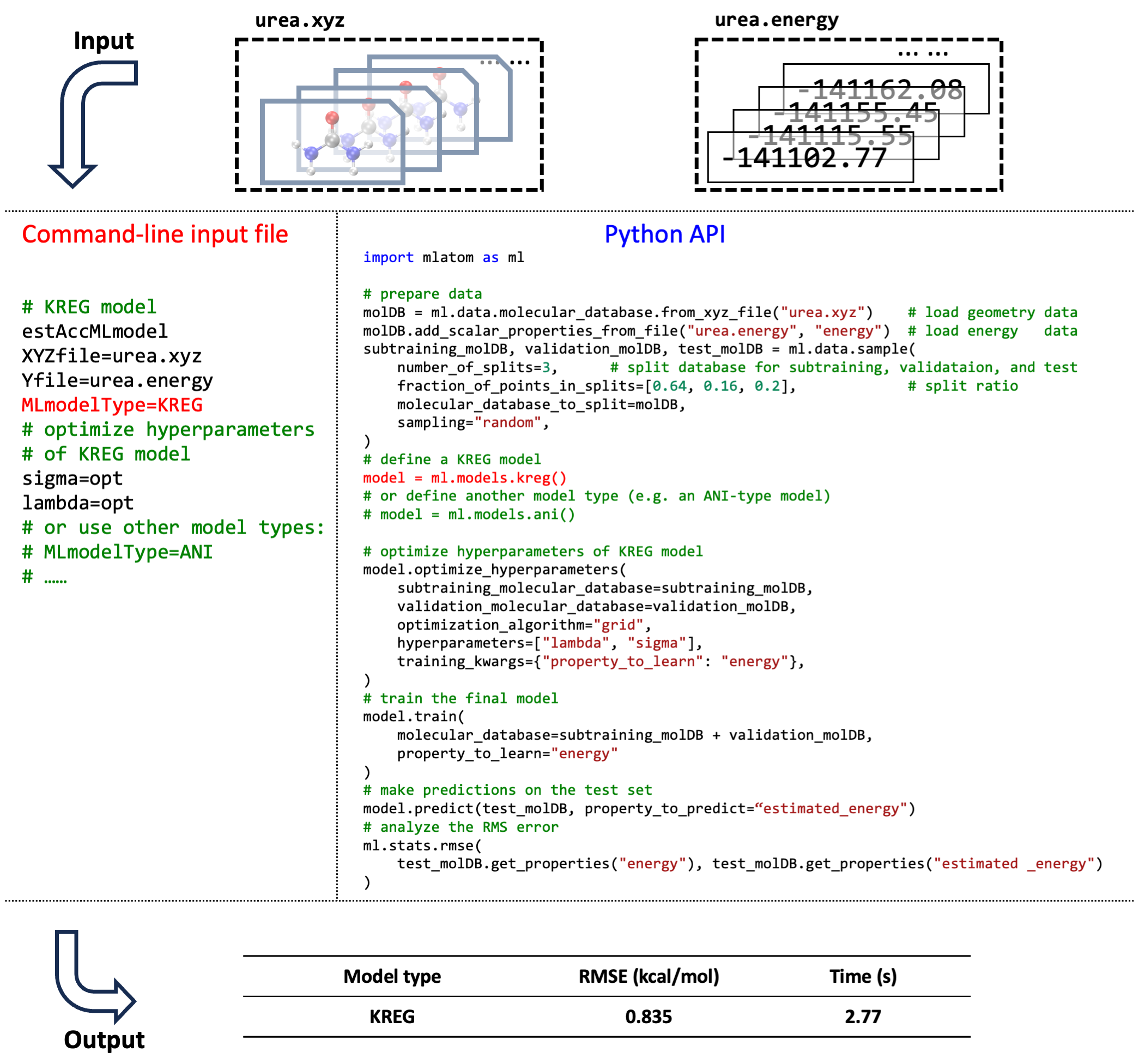
另外还显示了KREG模型所需的超参数优化。KREG模型训练速度快且准确(在几秒钟内达到均方根误差低于1 kcal/mol),这是对于小型分子数据库而言的典型情况,而对于较大数据库,可能更倾向于使用基于NN的模型。图中还以注释形式显示了使用不同类型的ML模型(例如ANI类型)的命令行和Python脚本输入。
参见 MACE 模型教程。
设计和训练自定义ML模型
MLatom的用户还可以使用各种KRR核函数在任何输入向量和标签集上创建模型。在这种情况下,强烈建议进行超参数优化。在所有其他方面,训练这些KRR模型与训练预定义模型类似,即通过将数据集拆分为训练和验证所需的子集来准备数据集。
重要的是,用户可以使用模型树构建各种复杂性的模型。这样的复合模型的特殊情况包括Δ-learning和自校正模型,它们可以通过提供输入向量或XYZ坐标和标签来进行类似于其他ML模型的训练。在Δ-learning的情况下,用户必须提供基线值。对于其他更复杂的模型,用户必须单独训练、组合每个组件。
超参数优化
ML模型的性能强烈依赖于选择的超参数,例如用于训练核方法的正则化参数和神经网络中的层数。因此,通常需要优化超参数以获得合理的结果并提高准确性。超参数优化通常需要多次训练,这使其成为一项昂贵的工作,必须谨慎平衡性能/成本问题。
MLatom可以通过使用许多可用算法中的一种最小化验证损失来优化超参数。验证损失通常基于验证集中的错误,该错误可以是单个延迟验证集,也可以是组合的交叉验证错误。
对于少数几个超参数,可以使用对数或线性刻度上的鲁棒网格搜索来找到最佳值。这是核方法的常见选择(见上面优化KREG模型超参数的示例,该模型是核方法)。对于更多的超参数,建议使用其他算法。流行的选择包括具有树形Parzen估计器(TPE)的Bayesian优化和多种SciPy优化器。
验证损失的选择也很重要。在大多数情况下,MLatom会最小化带标签数据的均方根误差(RMSE)。然而,当提供多个标签时,即用于学习PES的能量和能量梯度,应根据如何在验证损失中组合它们来选择。默认情况下,MLatom计算能量和梯度的RMSE的几何平均值。用户还可以选择RMSE的加权和,但在这种情况下,必须选择权重。此外,用户可以向MLatom提供任何自定义的验证损失函数,它可以是任意复杂的。
评估模型
一旦模型被训练,通常将其部署到生产模拟之前,需要评估其泛化能力。MLatom为这样的评估提供了专用选项。最简单且最广泛使用的方法之一是计算独立保留测试集的误差,该测试集未在训练中使用。需要强调的是,在MLatom的术语中,测试集与训练集没有重叠,后者可能包括子训练集和验证子集。另外,如果在计算上可负担得起时,推荐使用交叉验证及其变体留一交叉验证,尤其是对于小数据集。MLatom为测试集提供了广泛的误差度量,包括RMSE、平均绝对误差(MAE)、平均带符号误差、Pearson相关系数、R2值、异常值等。对于大多数模型,包括Δ-learning和自校正模型,可以进行带有训练和超参数优化的测试。
由于误差取决于训练集的大小,比较不同模型时,体现这种依赖性的学习曲线非常有用。MLatom可以生成学习曲线,这在制定选择ML原子间势的指南时发挥了重要作用。
请参阅关于如何对不同类型的机器学习潜力进行基准测试的专用 教程 (由于没有显示Python API的使用,因此有点过时)。
MLatom的开发和贡献
对于开发人员,MLatom提供了一个灵活的平台,用于实现新的接口,因为只需提供支持使用新模型进行预测(训练)的新类。例如,MACE的实现在一天内完成,测试需要额外一天的时间。一旦实现了这些模型,它们就可以轻松用于模拟。
欢迎向MLatom的主要GitHub存储库(https://github.com/dralgroup/mlatom)贡献,并可以通过分支(根据要求)和贡献者可能为其方法和功能的私有开发创建的分支进行拉取请求。经过主要开发团队的审查和可能的调整后,拉取请求可能会合并到官方版本中。
支持与联系
如果您有进一步的问题、批评和建议,我们很乐意通过 电子邮件 、 Slack (首选)或微信(请发送电子邮件请求将您添加到XACS用户支持组)以英文或中文接收您的意见。