单点能计算
这篇教程展示了如何用MLatom计算单点能。
MLatom能够使用各种模型和方法计算单点能,包括:
量子力学方法,
混合量子力学/机器学习方法,
预训练的机器学习模型和用户定义的机器学习模型。
有关MLatom中模型的更多详细信息,请查看 这篇概览 我们也在Youtube上传了本节教程的 视频 。
在这里,我们将说明如何使用MLatom中不同类别的方法计算 ISOL24 数据集中糖的异构化能
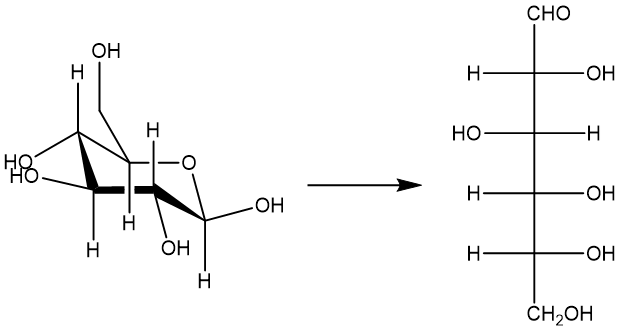
您可以下载所需的文件 sugar_iso.xyz ,其中包含两个异构体的几何结构,格式为XYZ(以埃为单位)。这里的基本思路是我们分别计算异构体的单点能量并计算它们的能量差异。由于MLatom是一个面向数据的软件包,它本质上支持多分子的计算。我们首先展示如何通过输入文件/命令行选项进行计算,然后展示 如何在Python中获取单点性质
使用量子力学方法计算单点能
MLatom支持使用各种流行的程序进行单点量子力学计算,如 Gaussian , PySCF , xTB 等。有关使用量子力学方法进行模拟的详细手册,请参阅 使用量子力学方法模拟 。
进行单点能量计算的典型输入文件应包含定义几何结构的 xyzfile 和定义存储能量的文件的 yestfile 。对于量子力学方法,用户需要提供方法 method 和相应的量子力学程序 qmprog 。以下是在Gaussian中进行B3LYP/6-31G*水平的能量计算的输入文件示例:
method=B3LYP/6-31G*
qmprog=gaussian
xyzfile=sugar_iso.xyz
yestfile=enest.dat
通过运行 $mlatom mlatom.inp > mlatom.out ,您将会得到如下MLatom输出文件:
******************************************************************************
Energy of molecule 1: -687.1484077000000 Hartree
Energy of molecule 2: -687.1383648000000 Hartree
==============================================================================
Wall-clock time: 83.08 s (1.38 min, 0.02 hours)
MLatom terminated on 03.02.2024 at 09:35:57
==============================================================================
在输出文件中,两个分子的能量的排列顺序与输入 .xyz 文件的分子顺序一致。
使用混合量子力学/机器学习方法计算单点能
混合QM/ML方法的成功例子之一便是 AIQM1 它是一种通用的方法,能够以较低级别的半经验量子力学方法的代价逼近金标准的耦合簇方法。它利用了差值学习策略的优势,通过最新的ML模型集合来纠正不太准确的半经验方法(见下图)。此外,在MLatom中,用户还可以定义自己的差值学习模型,目前可以通过PythonAPI轻松实现(请参阅 Python API教程 )
对于AIQM1,用户只需要在输入文件第一行要求进行 AIQM1 计算:
AIQM1 # 1. request AIQM1 method
xyzfile=sugar_iso.xyz # 2. file with geometry
yestfile=enest.dat # 3. file to store energy
在运行 $mlatom mlatom.inp > mlatom.out 之后,用户会得到如下 mlatom.out :
******************************************************************************
Properties of molecule 1
Standard deviation of NN contribution : 0.00094895 Hartree 0.59548 kcal/mol
NN contribution : 0.00275644 Hartree
Sum of atomic self energies : -578.89384843 Hartree
ODM2* contribution : -107.70671483 Hartree
D4 contribution : -0.00358197 Hartree
Total energy : -686.60138879 Hartree
Properties of molecule 2
Standard deviation of NN contribution : 0.00066560 Hartree 0.41767 kcal/mol
NN contribution : 0.00159327 Hartree
Sum of atomic self energies : -578.89384843 Hartree
ODM2* contribution : -107.68954498 Hartree
D4 contribution : -0.00351388 Hartree
Total energy : -686.58531402 Hartree
==============================================================================
Wall-clock time: 0.77 s (0.01 min, 0.00 hours)
MLatom terminated on 03.02.2024 at 09:20:02
==============================================================================
对于每个分子,AIQM1方法中所有的能量成分都列在了输出文件中,包括ODM2*能量贡献 ODM2* energy , D4色散校正 D4 enengy 和神经网络能量贡献( NN contribution 和 atomic self energies 之和)。 standard deviation of NN contribution 即神经网络能量间的标准偏差,可以用来判断AIQM1是否可信。
作为对比,我们在下表中列出了每种方法计算的异构化能以及它们对应的计算时间:
Method |
\(\Delta E_\text{r}\) (kcal/mol) |
time (s) |
|---|---|---|
reference |
10.69 |
- |
AIQM1 |
10.08 |
0.77 |
B3LYP/6-31G* |
6.27 |
83 |
使用用户训练的机器学习方法计算单点能
MLatom可以从文件中读取用户训练的模型,并用它对新数据进行预测(以输入向量X或XYZ坐标的形式给出)。用户应提供 MLmodelType 和/或 MLprog 参数。下表列出了可用的机器学习模型类型和使用的程序:
MLmodelType |
MLporg |
|---|---|
KREG |
MLatomF |
sGDML |
sGDML |
GAP-SOAP |
GAP |
PhysNet |
PhysNet |
DeePot-SE |
DeepMD-kit |
ANI |
TorchANI |
MACE |
MACE |
我们将使用预训练的 MACE 模型预测分子能量。可以在此处下载示例的MACE模型文件 mace.pt 。以下是如何使用用户训练的MACE模型进行单点能计算的示例:
useMLmodel # 1. requests usage of ML model for prediction
MLmodelType=MACE # 2. request the MACE model
MLmodelIn=mace.pt # 3. the file with the trained model should be provided, here it is mace.pt file
xyzfile=sugar_iso.xyz # 4. file with geometries
对于如何使用MLatom训练机器学习模型,请参照 MLatom教程
梯度和黑塞矩阵
如果用户需要梯度和黑塞矩阵,可以在输入文件中加入两行:
AIQM1 # 1. request AIQM1 method
xyzfile=sugar_iso.xyz # 2. file with geometry
yestfile=enest.dat # 3. file to store energy
ygradxyzestfile=grad.dat # 4. file to store gradients
hessianestfile=hess.dat # 5. file to store hessian
ygradxyzestfile 会创建储存梯度的文件, hessianestfile 会创建储存黑塞矩阵的文件。这些关键词大小写不敏感,但定义文件名的关键词需要小写。
电荷和多重度
如果分子具有除默认电荷(默认为0)和多重度(默认为1)以外的其他值,输入文件可以如下所示
method=B3LYP/6-31G*
qmprog=gaussian
xyzfile=sp.xyz
yestfile=enest.dat
charges=0,1
multiplicities=1,3
通过PyAPI进行计算
定义方法
Python API提供了MLatom方法更灵活的使用方式。详细介绍在 python API中的方法 部分中提供。对于QM方法、AIQM1和预训练的机器学习模型,用户可以使用 mlatom.models.methods 模块定义方法。这里有三个常用的参数
method:定义您想用的方法program:该方法需要的程序nthreads:程序使用的线程数
比如,您可以按如下定义QM方法
method = mlatom.models.methods(method='b3lyp/6-31G*', program='gaussian')
# method = mlatom.models.methods(method='b3lyp/6-31G*', program='pyscf', nthreads=18)
备注
AIQM1使用 qm_program 关键词来定义ODM2*计算使用的程序
加载用户训练的模型需要使用下表中不同的模块。有关这些模块的详细信息,请参阅 API手册
model type |
module |
|---|---|
KREG |
|
sGDML |
|
GAP-SOAP |
|
PhysNet |
|
DeePot-SE |
|
ANI |
|
MACE |
|
使用定义的方法进行计算
在这里,我们将介绍使用Python API计算糖异构化能的流程。MLatom中使用的能量单位是Hartree。由于计算涉及多个分子,我们从使用 mlatom.data.molecular_database 类加载这些分子开始,然后使用定义的方法中的 predict 函数执行计算
import mlatom as ml
# read molecule from .xyz file
molDB = ml.data.molecular_database.from_xyz_file('sugar_iso.xyz')
# define method
model = ml.models.methods(method='AIQM1')
# model = ml.models.methods(method='B3LYP/6-31G*'. program='pyscf') ## calculations using QM methods
model.predict(molecular_database=molDB)
print(molDB[0].energy)
print(molDB[1].energy)
print(molDB[1].energy-molDB[0].energy)
如果需要梯度和黑塞矩阵,用户可以通过以下方式进行计算
model.predict(molecular_database=molDB, calculate_energy_gradients=True, calculate_hessian=True)
如果只使用一个分子,用户也可以使用 mlatom.data.molecule 类加载分子
mol = ml.data.molecule.from_xyz_file('sp.xyz')
并且使用它进行计算
model.predict(molecule=mol)
用户还可以通过以下方式为 molecular_database 或 molecule 定义电荷和多重度:
mol.charge = 1
mol.multiplicity = 3
molDB.charges = [1,1]
molDB.multiplicities = [1,3]
获取性质
在MLatom中,能量和黑塞矩阵将存储在 molecule 对象中,而梯度将存储在 atom 对象中(详细信息请参阅 Python API中的数据 部分)。用户可以通过以下方式获取它们:
mol.energy
mol.get_energy_gradients()
mol.hessian
molecular_database 提供了 get_properties 函数来从每个 molecule 对象中提取性质。用户可以用以下方式获取梯度和黑塞矩阵的数组:
molDB.get_properties('energy')
molDB.get_xyz_vectorial_properties('energy_gradients')
molDB.get_properties('hessian')
使用用户自定义的混合模型计算单点能
MLatom通过使用 mlatom.models.model_tree_node 模块具有将不同模型组合在一起的能力,其工作方式类似于树结构(详细信息请参阅 Python API中的模型树节点 )
这里我们以自定义的差值学习模型为例,该模型是根据H2的全组态相互作用和Hartree–Fock能量之间的差异进行训练的。预训练的模型文件可以从这里下载 delta_FCI-HF_h2_ani.npz
你可以按如下方式定义2个子节点:
baseline=ml.models.model_tree_node(name='baseline', operator='predict', model=ml.models.methods(method='HF/STO-3G', program='pyscf')
delta_correction=ml.models.model_tree_node(name='delta_correction', operator='predict', model=ml.models.ani(model_file='delta_FCI-HF_h2_ani.npz')`
您可以看到有一个 operator 对您定义的模型进行操作(这里每个模型只执行预测任务)。现在我们定义父节点为:
ml.models.model_tree_node(name='hybrid', operator='sum', children=[baseline, delta_correction])
在这里, operator 更改为 sum ,因为我们将在此节点上执行的操作是将两个子节点的预测值相加。
最后,我们将这些部分组合在一起并预测H2的能量。您可以在此处下载所需的几何文件 h2_init.xyz
import mlatom as ml
# Get the initial guess for the molecules to optimize
mol = ml.data.molecule.from_xyz_file('h2_init.xyz')
# Let's build our own delta-model
# First, define the baseline QM method, e.g.,
baseline = ml.models.model_tree_node(name='baseline', operator='predict', model=ml.models.methods(method='HF/STO-3G', program='pyscf'))
# Second, define the correcting ML model, in this case the KREG model trained on the differences between full CI and HF
delta_correction = ml.models.model_tree_node(name='delta_correction', operator='predict', model=ml.models.ani(model_file='delta_FCI-HF_h2_ani.npz'))
# Third, build the delta-model which would sum up the predictions by the baseline (HF) with the correction from ML
delta_model = ml.models.model_tree_node(name='delta_model', children=[baseline, delta_correction], operator='sum')
delta_model.predict(molecule=mol)
print(mol.energy)
问题或建议
If you have further questions, criticism, and suggestions, we would be happy to receive them in English or Chinese via email, Slack (preferred), or QQ (135258964). See our contact page for more information on how to subscribe to updates and follow us on social media.