从分子动力学计算振动光谱
本教程将展示如何从 分子动力学 轨迹计算振动光谱(红外光谱和功率谱)。
我们还制作了该教程的视频,你可以在 bilibili 上找到它。
理论
红外光谱可以通过对偶极矩的自相关函数做傅里叶变换得到:
\(P(\nu)\propto\frac{\nu\text{tanh}(h\nu/2k_{\text{B}}T)}{n(\nu)}\int\langle\mathbf{\mu}(\tau)\mathbf{\mu}(t+\tau) \rangle _{\tau}e^{-i2\pi\nu t} \text{d}t\),
其中, \(\langle \mathbf{\mu}(\tau)\mathbf{\mu}(t+\tau) \rangle _{\tau}\) 是偶极矩的自相关函数, \(\nu\) 是频率, \(h\) 是普朗克常数, \(k_{\text{B}}\) 是玻尔兹曼常数, \(T\) 是温度, \(n(\nu)\) 是折射率,在气相中,该值为1。在做快速傅里叶变换之前,我们会先用Hann窗口函数处理自相关函数,用户可以调节自相关深度(autocorrelation depth)来控制谱峰的宽度。用户也可以在自相关函数后用零值填充(zero padding)来提高谱图的精度。
类似地,功率谱也可以从速度的自相关函数得到:
\(P(\nu)\propto\int \langle\mathbf{v}(\tau)\mathbf{v}(t+\tau)\rangle_{\tau}e^{-i2\pi\nu t} \text{d}t\),
其中, \(\langle\mathbf{v}(\tau)\mathbf{v}(t+\tau)\rangle_{\tau}\) 是速度的自相关函数。
从分子动力学轨迹计算振动光谱的理论部分可以参考以下两篇文章: DOI: 10.1039/C3CP44302G 和 DOI: 10.1039/C6CP05849C 。
MLatom中的计算细节可以参考 这篇文章(DOI: 10.1039/D3CP03515H) 。使用此功能时请引用该文章
备注
MLatom中的一些QM方法(例如使用Gaussian和MNDO程序的方法)会在用户计算单点能时自动提供偶极矩。除了AIQM1,其它所有的预训练机器学习模型都不能提供偶极矩。AIQM1提供的是ODM2*级别的偶极矩,且只有当使用MNDO(而非Sparrow)时有效。所以在计算红外光谱前请确认使用的方法是否能提供偶极矩。所有的方法都能用来计算功率谱,因为其只需要速度即可得到。
使用输入文件/命令行
接下来我们将展示如何通过输入文件或命令行计算振动光谱。输入文件中展示了如何从h5md格式的分子动力学轨迹计算乙醇的红外光谱。在这里我们提供了乙醇的轨迹,它是用AIQM1在300K的Nose-Hoover温标下得到的。轨迹的步长为0.5飞秒。分子动力学轨迹可以在这里下载: ethanol_traj.h5 (h5md格式), ethanol_traj.xyz (xyz坐标), ethanol_traj.vxyz (xyz速度)和 ethanol_traj.dp (偶极矩)。
MD2vibr # 1. requests vibrational spectra from MD trajectory
trajH5MDin=ethanol_traj.h5 # 2. file with MD trajectory in h5md format
dt=0.5 # 3. time step of trajectory; unit: fs
start_time=0 # 4. use trajectory starting from 0 fs
end_time=10000 # 5. use trajectory ending at 10000 fs
autocorrelationDepth=1024 # 6. autocorrelation depth; unit: fs
zeropadding=1024 # 7. zero padding length; unit: fs
output=ir # 8. output infrared spectrum
你会在文件夹里找到两个文件: ir.png 和 ir.npy 。前者是红外光谱的图片,后者是谱图的numpy数组。计算得到的红外光谱如下所示:
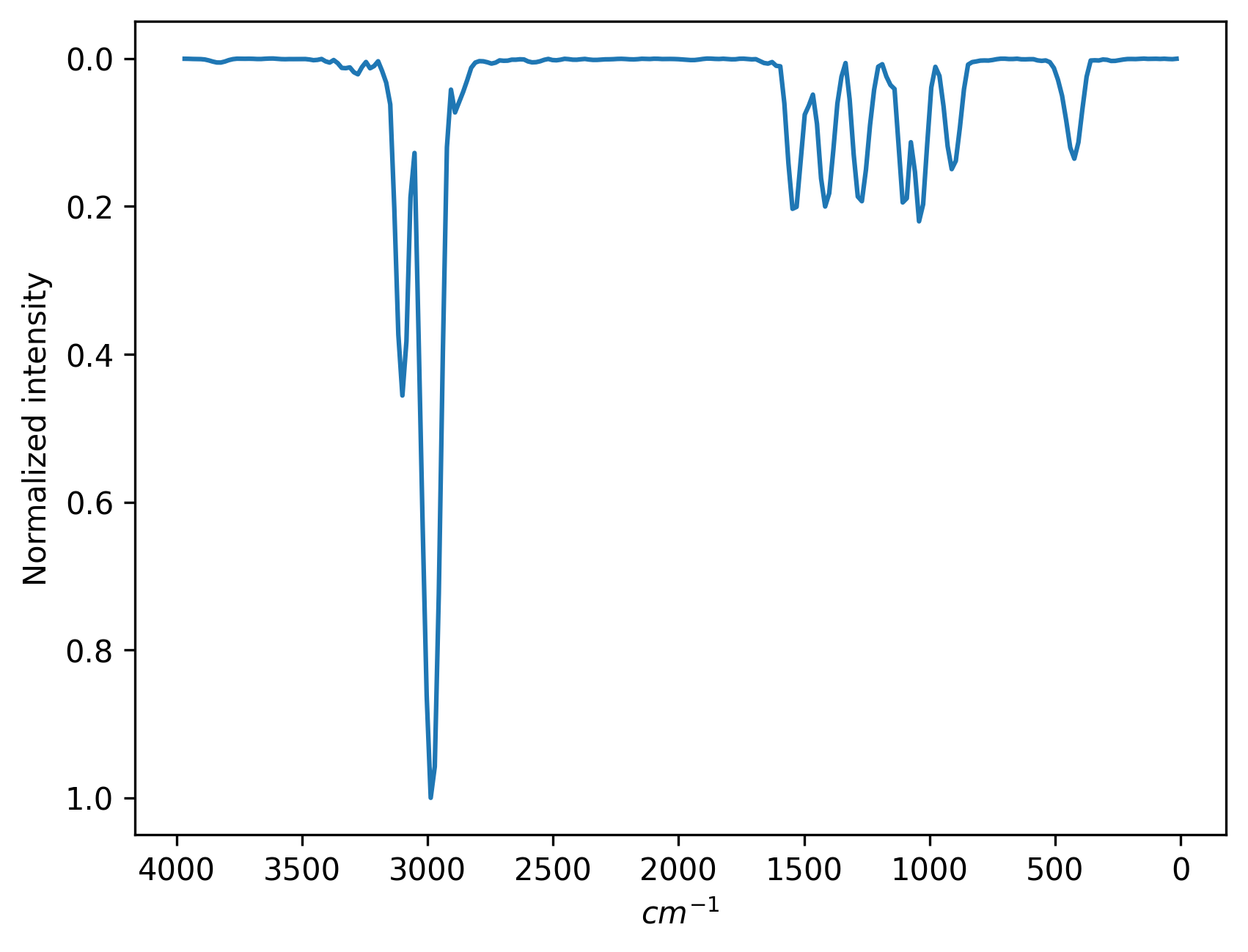
关键词 output 可以用来控制计算哪种振动光谱。 output=ir 计算红外光谱, output=ps 计算功率谱。下面是计算功率谱的输入文件。
MD2vibr # 1. requests vibrational spectra from MD trajectory
trajH5MDin=ethanol_traj.h5 # 2. file with MD trajectory in h5md format
dt=0.5 # 3. time step of trajectory; unit: fs
start_time=0 # 4. use trajectory starting from 0 fs
end_time=10000 # 5. use trajectory ending at 10000 fs
autocorrelationDepth=1024 # 6. autocorrelation depth; unit: fs
zeropadding=1024 # 7. zero padding length; unit: fs
output=ps # 8. output power spectrum
计算得到的功率谱如下所示:
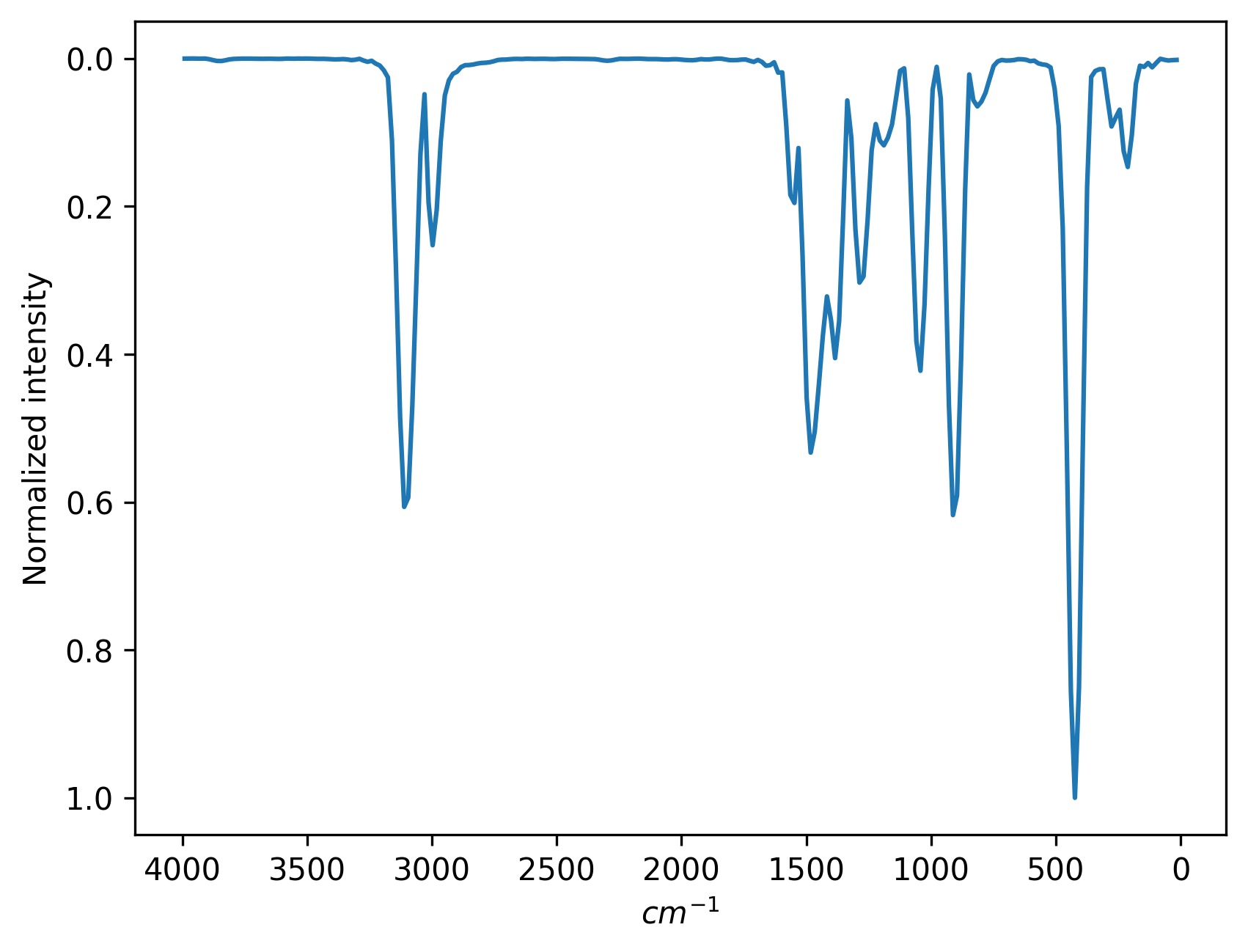
除了h5md格式的轨迹,还可以直接提供偶极矩(用来计算红外光谱)和速度(用来计算功率谱)的文本文件,需要使用 trajdpin 或 trajVXYZin 关键词。下面是两种情况的例子。
MD2vibr # 1. requests vibrational spectra from MD trajectory
trajdpin=ethanol_traj.dp # 2. file with dipole moments in plain text
dt=0.5 # 3. time step of trajectory; unit: fs
start_time=0 # 4. use trajectory starting from 0 fs
end_time=10000 # 5. use trajectory ending at 10000 fs
autocorrelationDepth=1024 # 6. autocorrelation depth; unit: fs
zeropadding=1024 # 7. zero padding length; unit: fs
output=ir # 8. output infrared spectrum
MD2vibr # 1. requests vibrational spectra from MD trajectory
trajVXYZin=ethanol_traj.vxyz # 2. file with velocities in plain text
dt=0.5 # 3. time step of trajectory; unit: fs
start_time=0 # 4. use trajectory starting from 0 fs
end_time=10000 # 5. use trajectory ending at 10000 fs
autocorrelationDepth=1024 # 6. autocorrelation depth; unit: fs
zeropadding=1024 # 7. zero padding length; unit: fs
output=ps # 8. output power spectrum
备注
原则上我们推荐使用AIQM1方法。通常它在一定的计算代价下是最准确的。AIQM1的偶极矩是ODM2*级别的。但是偶极矩只有在使用MNDO程序时才能获得。我们推荐安装MNDO以解锁AIQM1的全部功能。XACS云计算平台使用的是Sparrow,目前不提供偶极矩,而且它只提供数值梯度,这限制了它在MD中的应用。AIQM1的另一个限制是它只能用于CHNO元素。ANI-1ccx是XACS平台上一个好的替代方案,但是它同样不能计算偶极矩,而且它只能用于计算中性闭壳层的基态体系。
其它关键词
关键词 start_time 和 end_time 可以控制选择轨迹的某一部分计算谱图。默认情况下,MLatom会着用整条轨迹。 autocorrelationDepth 控制自相关深度,它会影响峰的宽度; zeropadding 控制零填充的长度,它会影响谱图的分辨率。这两个设置的默认值是1024飞秒。MLatom会用谱图的最大值对谱图进行归一化。如果想要获得原始谱图,可以使用 normalize_intensity=0 。
使用API
接下来将展示如何用MLatom的API计算振动光谱。
import mlatom as ml
# Loading MD trajectory in h5md format
traj = ml.data.molecular_trajectory()
traj.load(filename='ethanol_traj.h5',format='h5md')
moldb = ml.data.molecular_database()
moldb.molecules = [step.molecule for step in traj.steps]
# Loading MD trajectory in plain text
# moldb = ml.data.molecular_database.from_xyz_file('ethanol_traj.xyz')
# moldb.add_xyz_vectorial_properties_from_file('ethanol_traj.vxyz','xyz_velocities')
# with open('ethanol_traj.dp','r') as f:
# lines = f.readlines()
# for ii in range(len(moldb)):
# moldb[ii].dipole_moment = [float(each) for each in lines[ii].strip().split()]
# Calculate vibrational spectra
vibr = ml.vibrational_spectrum(molecular_database=moldb,dt=0.5)
# Infrared spectrum
freqs_ir,ints_ir = vibr.plot_infrared_spectrum(filename='ethanol_ir.png', # file to save the figure
autocorrelation_depth=1024, # autocorrelation depth; unit: fs
zero_padding=1024, # zero padding; unit: fs
lb=0, # start time; unit: fs
ub=10000, # end time; unit: fs
normalize=True, # normalize the spectrum
return_spectrum=True, # get the spectrum array from the function
format='modulus') # use the modulus of the dipole moments
# Power spectrum
freqs_ps,ints_ps = vibr.plot_power_spectrum(filename='ethanol_ps.png', # file to save the figure
autocorrelation_depth=1024, # autocorrelation depth; unit: fs
zero_padding=1024, # zero padding; unit: fs
lb=0, # start time; unit: fs
ub=10000, # end time; unit: fs
normalize=True, # normalize the spectrum
return_spectrum=True) # use the modulus of the dipole moments
# Save the spectra
np.save('ethanol_ir_saved.npy',np.array((freqs_ir,ints_ir)))
np.save('ethanol_ps_saved.npy',np.array((freqs_ps,ints_ps)))
关键词与输入文件有些许不同。 lb 和 ub 可以控制轨迹的开始时间和结束时间。 autocorrelation_depth 和 zero_padding 分别控制自相关深度和零填充长度。 normalize=True 会让谱图归一化。使用 return_spectrum=True 可以让函数返回图谱的数组。在红外光谱中,还可以使用 format='modulus' 或 format='vector' 来选择使用偶极矩的模或XYZ向量。
问题或建议
If you have further questions, criticism, and suggestions, we would be happy to receive them in English or Chinese via email, Slack (preferred), or QQ (135258964). See our contact page for more information on how to subscribe to updates and follow us on social media.