6. Molecular dynamics
Molecular dynamics (MD) is the field where ML is most widely used and ML successes in MD stimulated the wider adoption of ML in chemistry.
6.1. Slides
6.2. What is MD and why is it important?
[Credit: Fuchun Ge, ethanol MD]
Atoms in molecules are always in constant motion and, hence, to properly simulate molecules, we need to take this into account. The most widely used approach for directly simulating atomic motions is molecular dynamics (MD). By performing MD we can get chemically intuitive view on the processes on the atomistic level and derive spectral and thermochemical properties. We can also better understand how the chemical reactions happen as they are rearrangement of atoms in space and time.
But how can we simulate the atomic motion? In principle, we need to solve the time-dependent Schrodinger equation for both electrons and nuclei. This is, unfortunately, numerically not easy. In MD, we make a huge simplification by asssuming that the atoms move according to the classical Newton’s law and then working our way from the Neston’s laws of motion. It means that we need to solve the equations of motion starting with the basic equation:
Now, given the known initial conditions (positions of atoms and their velocities), we can calculate the position of atoms in a short time increment (time step) and repeat this procedure until we propagate the nuclear motion for as long as we need. The obtained sequence of geometries and associated properties is called MD trajectory. The velocity Verlet algorithm is a popular but not the only way to do this. To give a better idea how MD is is propagated, you can check the following flowchart:
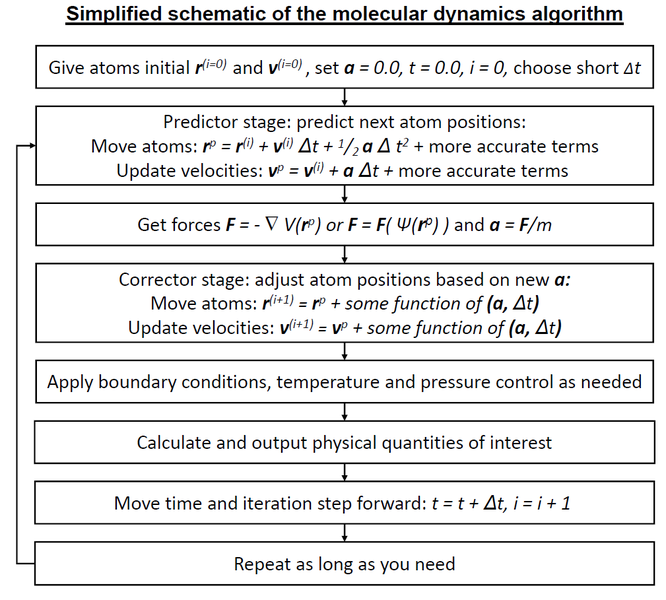
[Credit: Knordlun, CC BY-SA 3.0, via Wikimedia Commons.]
The key component in above MD algorithm is calculation of forces. They can be obtained from the molecular mechanics force fields (not covered in this course) and QM calculations (and then such MD is called Born–Oppenheimer MD because the forces are calculated for static nuclei within the Born–Oppenheimer approximation). The forces are just negative energy gradients, i.e., negative derivatives of potential energy with respect to nuclear coordinates:
Let’s see MD in action:
Example MD-1.
Propagate an MD trajectory of H2 molecule with B3LYP/6-31G*.
Visualize the trajectory. What is the range the bond length changes (min, max)?
How much time did it take to complete the calculations?
The MLatom@XACS input file:
MD
B3LYP/6-31G*
dt=0.5 # fs - time step
trun=500 # fs - propagation duration
initConditions=random
initXYZ='2
H 0 0 0
H 0 0 0.8'
6.3. Why is molecular dynamics expensive?
From the above algorithm it is obvious that MD is quite an expensive business. Even for H2, where DFT was very fast, it takes some time to propagate a relatively short trajectory and, often, we need to propagate many trajectories for a longer time! Think about the typical reaction rates and how long would you need to propagate MD to see reaction events…
There is also no way to parallelize the calculation of the time steps – they should be run one after another in the strict order, further limiting the speed of MD. In addition, to obtain better quality of MD you need smaller time steps – but not too small as the calculation time will increase correspondingly and numerical errors will accumulate faster too.
6.4. How can ML accelerate MD?
Given the high cost of MD, it is highly desirable to find a way to accelerate it with ML because of its high speed.
How would you proceed to use ML for accelerating MD?
6.4.1. Accelerating calculation of forces
The obvious way is to accelerate the slowest part of MD which is calculation of forces. We have to evaluate them at each time step, e.g., with expensive QM methods in case of Born–Oppenheimer MD. Indeed, using ML for accelerating the forces evaluation is one of the most widely used applications of MD in computational chemistry (and probably in chemistry in general). How can we calculate forces with ML? One way to do it is to directly learn the forces themselves and, indeed, some works doing exactly this. However, another, more established approach, is to use ML potentials which, as we know from ML-PES lecture, are energy functions of nuclear coordinates and, hence, ML can be viewed as surrogate models to QM. ML models are also analytical functions and given automatic differentiation capabilities of many ML packages, it is straightforward to obtain the first-order derivatives and forces from such potentials. The calculations will be much faster as the result. Let’s the use of an MLP in action.
Example MD-2.
Propagate an MD trajectory of H2 molecule with ANI-1ccx.
Visualize the trajectory. What is the range the bond length changes (min, max)?
How much time did it take to complete the calculations?
Compare time to DFT calculations.
Does ANI-1ccx produce physically-meaningful trajectory?
MD
ANI-1ccx
dt=0.5 # fs - time step
trun=500 # fs - propagation duration
initConditions=random
initXYZ='2
H 0 0 0
H 0 0 0.8'
Note the speed up for the hydrogen molecule is not so dramatic, but try something bigger!
Now, if you analyzed the trajectory carefully enoough you would notice how ANI-1ccx produces completely unphysical trajectory, where the atoms ‘tunnel’ through each other. This is the result of a known problem with ANI-1ccx which has wrong potential energy curve for the hydrogen molecule.
ANI-1ccx has two minima on PES (which you should remember from the previous homework analysis) and the energy for small internuclear distance is not going exponentially up as it should due to the repulsion of nuclei! This is ML and anything can happen – it is based on what it saw in the data and has no physical model restraining it from behaving unphysically. If you also noticed, at large distance the potential energy drops while it should not (it should plateou). This is unfortunately also a common behavior or MLP which has a consequence that many MDs with MLPs lead to unphysical explosion of molecules.
Luckily, you can use your own ANI models trained on the FCI data! Let’s see how they perform:
Example MD-3.
Propagate an MD trajectory of H2 molecule with your own ANI model trained on FCI data.
Visualize the trajectory. What is the range the bond length changes (min, max)?
How much time did it take to complete the calculations?
Does this ANI model produce physically-meaningful trajectory?
Modify the input file from previous task as needed.
Finally, you have obtained a fast dynamics which also looks reasonable.
6.4.2. Directly learning dynamics
Why not use ML to directly learn the nuclear positions as a function of time? Indeed, we were the first to suggest and demonstrate the feasibility of such a novel of way of accelerating dynamics with ML. In this approach, we seek a function
Currently, we are working on a generalization of our previous ML model so that it can be used easier.
6.5. MD types
6.5.1. Constant energy vs constant temperature
MD can be propagated in many different modes. If we consider an isolated molecule, not interacting with an environment, then it should conserve total energy (some of the potential energy calculated with your QM or ML model and the kinetic energy of nuclei). You run such simulations in above tasks and they are said to be done in the NVE ensemble.
However, often, we want to simulate the process of a molecule in its environment without including all the environment (as it would be quite slow). Interacting with environment often means that the temperature on average should be more or less constant. You can do it with MLatom using the so-called NVT ensemble. There are many possible algorithms developed for the NVT ensemble called thermostats, e.g., Nosé–Hoover thermostat and Andersen which are implemented in MLatom. In this case, the total energy will not be conserved. You can see it for yourself in the corresponding example:
Example MD-4.
Propagate an MD trajectory of H2 molecule with your own ANI model trained on FCI data at 300 K using the NVT ensemble.
Plot how the total energy changes compared to the simulation in the NVE ensemble. What is the major difference?
Plot how the temperature changes too.
Is it expected?
You would need to add the following lines to your input file from previous task:
temperature=300
ensemble=NVT
thermostat=Nose-Hoover
Note
You can find total energy in the files traj.etot and temperature in the files traj.temp.
The key observation is that in the NVT ensemble the total energy is no longer conserved while in the NVE ensemble is does not deviate much. There is still noticable deviation of the total energy in the NVE ensemble. You can think about the reasons and how you can adjust the settings of the MD simulations to improve the energy conservation in the NVE simulations.
The temperature is, strictly speaking, not conserved in either of the simulation but in the NVT it varies narrower and the average should be closer to the target temperature.
Very often, in the NVT dynamics, the temperature rises or drops from the initial temperature to some more stable value with the average around the target temperature. Then you usually need to cut the initial (called equilibration) period before it becomes stable and then use for analysis the later part of the MD.
6.5.1.1. Energy conservation
In the NVE ensemble we want to conserve the total energy \(E_\text{tot}\) as the MD must adhere to the laws of physics. The total energy is a sum of the potential \(E_\text{pot}\) and kinetic \(E_\text{kin}\) energies. The potential energy is evaluated at the QM level or with MLP surrogate model as usual \(E_\text{pot} = f(\mathbf{R})\). The kinetic energy is a sum of the kinetic energies over nuclei:
All is good but for a small time step, most of MLPs will always produce an energy conserving MD which, as we know, can be still completely unphysical!
Hence, we suggested to evaluate the correctness of the trajectory by evaluating how much it deviates from the true energy conservation (i.e., for a dynamics with an exact but usually unknown potential) by evaluating the theoretical-best estimate of the total energy, where the potential energy is calculated for some snapshots from the trajectory at the highest affordable QM level, i.e.:
This criterion gives a much better error metric for the quality of MD.
6.5.2. Quasi-classical trajectory
Now think what will happen if you set the initial velocities to zero and start from the geometry with zero forces too? (What is the geometry with zero forces?). After you come to an answer, you can check it in the following exercise:
Example MD-5.
Start MD of H2 molecule with your own ANI model trained on FCI data from the optimized geometry (with the same model, you can take it from your previous exercise) using the NVE ensemble, and using the initial temperature of 0 K.
What happens to MD?
If you have done everything right, the simulation should lead to the trajectory where the atoms basically do not move. This is obvious if you consider how the MD is propagated - each steps changes the geometry based on the velocity and acceleration (defined by forces). Hence, for 0 K, velocity is zero by definition (kinetic energy should be zero). The molecule in the energy minimum has by definition energy derivatives (negative forces) equal to zero too, i.e., accelerations are zero. Hence, each next position must be the same as previous. You can still see small numerical noise.
The problem is that classical MD does not properly account for the fact that molecules are always in motion, i.e., they neglect zero-point vibrational energy (ZPE). In classical MD you can set the nuclear velocities so that their kinetic energy corresponds to the required initial instantenuous temperature. But this will definetely not reflect the ZPE.
In an attempt to recover the ZPE effect, you can propagate so-called quasi-classical trajectories (QCT). They differ from the classical ones by how the initial conditions are generated. To recover the ZPE, the initial geometries are sampled from the vibrational normal modes (typically calculated within the rigid-rotor harmonic-oscillator approximation) and the velocities are assigned so that the total energy (on average) includes ZPE. Then the QCT are propagated with the classical MD algorithm (e.g., velocity Verlet in MLatom).
There are several ways to generate initial conditions, with two popular ones implemented in MLatom:
Wigner sampling. Commonly used in surface-hopping dynamics.
sampling from the harmonic quantum Boltzmann distribution (called “thermal sampling” in the manual to its early implementation in the VENUS program). In contrast to Wigner sampling, it samples from higher vibrational levels. This sampling is commonly used for performing the downhill QCT investigation of transition states.
Now let’s run QCT trajectories:
Example MD-6.
Run quasi-classical trajectory MD of H2 molecule with your own ANI model trained on FCI data from the optimized geometry (with the same model, you can take it from your previous exercise).
For this, you first need to do freq calculations on the optimized geometry. These calculations will produce the freq1.json file which you can use in the following input file:
MD
MLmodelIn=energies_ani.pt
MLmodelType=ANI
dt=0.5 # fs - time step
trun=100 # fs - propagation duration
initTemperature=0
initConditions=Wigner
normalModeFile=freq1.json
How is this MD different to the previous one? What happens to MD?
The obvious qualitative difference is that even at 0 K, the QCT still has the motion of atoms in contrast to the classical MD. This is how it should be as the zero-point energy is always present in molecules, even at 0 K.
6.5.3. Other approaches you should know
Meta-dynamics. Even with all the ML acceleration, the time scales of typical chemical processes are much longer than what we can simulate. Hence, many approaches emerged which try to artificially accelerate the simulation so that at least the key dynamical features are recovered.
PIMD (path-integral molecular dynamics). QCT is one of the steps towards recovering at least ZPE but it is still not recovering quantum nuclear effects fully. A better approach is PIMD with its variants CMD and RPMD, which produces more robust and physically correct results but at a much higher cost. MLatom is now getting the PIMD implemented.
6.6. Further reading
A good introductory book about MD: D. Frenkel, B. Smit, J. Tobochnik, S. R. Mckay and W. Christian, Understanding Molecular Simulation, Elsevier, Bodmin, Cornwall, 1997.
Another book where you can read up about meta-dynamics and PIMD is X.-Z. Li, E. Wang, Computer Simulations of Molecules and Condensed Matter: From Electronic Structures to Molecular Dynamics. Peking University Press, 2014.